
Performer Miley Cyrus says that “Pink isn’t just a colour, it’s an attitude!”
Whether that is true or not is not for us to say.
What we can tell you is that ‘Pink’ is also a gene which is associated with Parkinson’s disease. And not just any form of Parkinson’s disease – people with early onset Parkinson’s (diagnosed before 40 years of age) often have specific mutations in this gene. And recently there has been new research published which may help these particular individuals.
Today’s post will review the new research and look at what it means for people with early onset Parkinson’s disease.

The actor Michael J Fox requires no introduction.
Especially in the Parkinson’s community where his Michael J Fox Foundation has revolutionised the funding and supporting of Parkinson’s disease research (INCREDIBLE FACT: Since 2000, The Michael J. Fox Foundation has funded more than US$450 MILLION of Parkinson’s disease research) and is leading the charge in the search for a cure for this condition.
Mr Fox has become one of the foremost figures in raising awareness about the disease that he himself was diagnosed with at just 29 years of age.
Wow, so young?
It is a common mistake to consider Parkinson’s disease a condition of the aged portion of society. While the average age of diagnosis floats around 65 years of age, it is only an average. The overall range of that extends a great distance in both directions.
Being diagnosed so young, Mr Fox would be considered to have early onset Parkinson’s disease.
What is early onset Parkinson’s disease?
Broadly speaking there are three basic divisions of Parkinson’s disease across different age ranges:
- Juvenile-onset Parkinson disease – onset before age 20 years
- Early-onset Parkinson disease – before age 50 years
- Late-onset Parkinson disease – after age 50 years is considered
The bulk of people with Parkinson’s disease are considered ‘late-onset’. The Juvenile-onset version of the condition, on the other hand, is extremely rare but cases do pop up regularly in the media (For example, click here). We have previously written about Juvenile-onset Parkinson disease (Click here for that post).
Early-onset Parkinson disease is more common than the juvenile form, but still only makes up a fraction of the overall Parkinsonian population. Some of those affected call themselves 1 in 20 as this is considered by some the ratio of early-onset Parkinson’s compared to late-onset.
How prevalent is early onset Parkinson’s?
In 2009, Parkinson’s UK published a report on the prevalence of Parkinson’s disease in the UK.
Using the General Practice Research Database (GPRD), which houses information about 7.2% of the UK population (or 3.4 million people in 2009), Parkinson’s UK found that the frequency of Parkinson’s disease in the general public was 27 cases in every 10,000 people (or 1 person in every 370 of the general population). The prevalence is higher in men (31 in every 10,000 compared to 24 in every 10,000 among females)
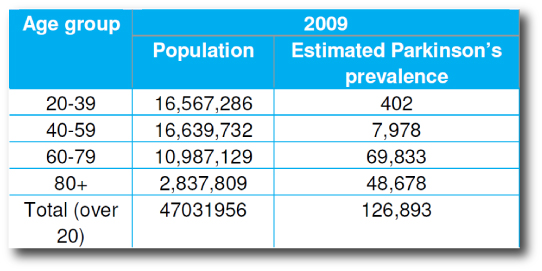
Source: ParkinsonsUK
As you can see from the table above, the number of people affected by early onset Parkinson’s disease is small when compared to the late-onset population.
Officially, the prevalence of early onset Parkinson’s in Europe is estimated to be 1 in every 8,000 people in the general population (Source: Orphanet). This makes the population of affected individuals approximately 5-10 % of all people with Parkinson’s. Hence the 1 in 20 label mentioned above.
Like older onset Parkinson’s, males are more affected than females (1.7 males to every 1 female case). In addition, women generally develop the disease two years later than men.
So what does ‘Pink’ have to do with early onset Parkinson’s?
First, let’s have a look at ‘Pink’ the gene.
PTEN-induced putative kinase 1 (or PINK1; also known as PARK6) is a gene that is thought to protect cells. Specifically, Pink1 is believed to interact with another Parkinson’s disease-associated protein called Parkin (also known as PARK2). Pink1 grabs Parkin and causes it to bind to dysfunctional mitochondria. Parkin then signals to the rest of the cell for that particular mitochondria to be disposed of. This is an essential part of the cell’s garbage disposal system.
Hang on a second. Remind me again: what are mitochondria?
Mitochondria are the power house of each cell. They keep the lights on. Without them, the lights go out and the cell dies.
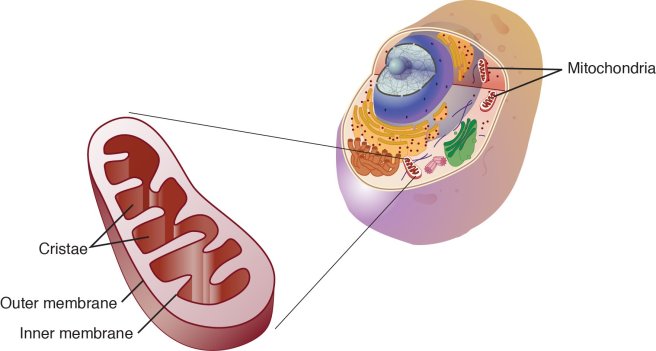
Mitochondria and their location in the cell. Source: NCBI
You may remember from high school biology class that mitochondria are bean-shaped objects within the cell. They convert energy from food into Adenosine Triphosphate (or ATP). ATP is the fuel which cells run on. Given their critical role in energy supply, mitochondria are plentiful and highly organised within the cell, being moved around to wherever they are needed.
When a cell is stressed by a toxic chemical, the organisation of mitochondria breaks down (as is shown in the image below, where everything except mitochondria (in green) and the nucleus (blue) has been made invisible:
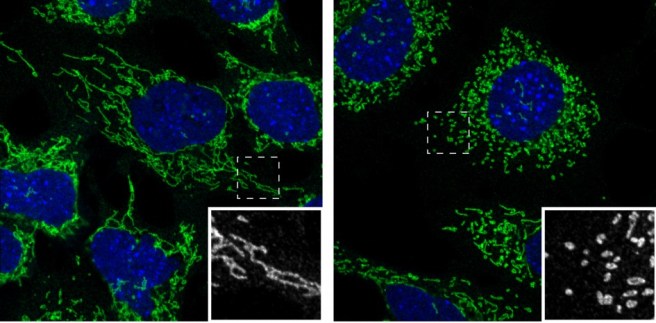
Mitochondria (green) in health cells (left) and in unhealthy cells (right).
The nucleus of the cell is in blue. Source: Salk Institute
In normal, healthy cells, PINK1 is absorbed by mitochondria and eventually degraded. In unhealthy cells, however, this process is inhibited and PINK1 starts to accumulate on the outer surface of the mitochondria. There, it starts grabbing the PARKIN protein. This pairing is a signal to the cell that this particular mitochondria is not healthy and needs to be removed.
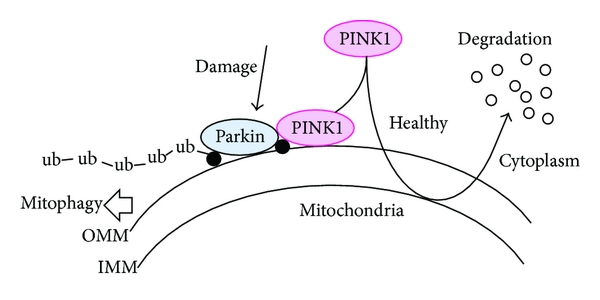
Pink1 and Parkin in normal (right) and unhealthy (left) situations. Source: Hindawi
The process by which mitochondria are removed is called autophagy. Autophagy is an absolutely essential function in a cell. Without it, old proteins will pile up making the cell sick and eventually it dies. Through the process of autophagy, the cell can break down the old protein, clearing the way for fresh new proteins to do their job.
Think of autophagy as the waste disposal process of the cell.
So why is Pink1 important to Parkinson’s disease?
In 2004 this research article was published:

Title: Hereditary early-onset Parkinson’s disease caused by mutations in PINK1
Authors: Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW.
Journal: Science. 2004 May 21;304(5674):1158-60.
PMID: 15087508
The researchers in this study were the first to report that mutations in the Pink1 gene were associated with increased risk of Parkinson’s disease. They found three families in Europe that exhibited a very similar kind of Parkinson’s disease and by analysing their DNA they determined that mutations in the Pink1 gene were directly linked to the condition.
They also looked at where in the cell Pink1 protein was located, noting the close contact with the mitochondria. In addition, they noted that the normal Pink1 protein provided the cell with protection against a toxic chemical, while the mutated version of Pink1 did not. These findings led the researchers to conclude that Pink1 and mitochondria may be involved in the underlying mechanisms of Parkinson’s disease.
And this initial study was quickly followed up 7 months later by this report:

Title: Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease.
Authors: Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, Reimsnider S, Tandon A, Hardy J, St George-Hyslop P, Singleton AB.
Journal: Arch Neurol. 2004 Dec;61(12):1898-904.
PMID: 15596610
In this study, the researchers analysed the Pink1 gene in 289 people with Parkinson’s disease and 80 neurologically normal control subjects. They identified 27 genetic variations, including a mutation in 2 unrelated early-onset Parkinson disease patients. They concluded that autosomal recessive mutations in PINK1 result in a rare form of early-onset Parkinson’s disease.
What does autosomal recessive mean?
Autosomal recessive means two copies of an abnormal gene must be present in order for the disease or trait to develop. That is to say, both parents will be carrying one copy of the mutation.
Mutations in the Pink1 gene have now been thoroughly analysed, with many mutations identified (the red and blue arrows in the image below). It is important, however, to understand that not all of those mutations are associated with Parkinson’s disease.
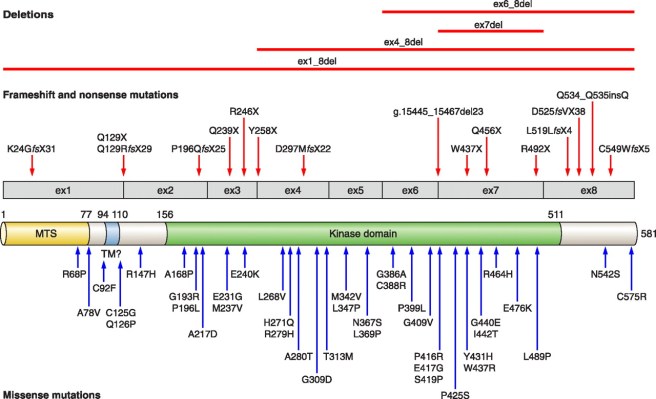
Looks complicated. Genetic variations in the Pink1 gene. Source: APS
So how do mutations in the Pink1 gene cause Parkinson’s disease?
We believe that the mutations in the Pink1 DNA result in malformed Pink1 protein. This results in Pink1 not being able to do what it is supposed to do. You will remember what we wrote above: Pink1 grabs Parkin when mitochondria get sick and Parkin signals for that mitochondria is be disposed of. Well, in the absence a properly functioning Pink1, we believe that there is a build up of sick mitochondria and this is what kills off the cell. All Parkinson’s disease-associated mutations in the Pink1 gene inhibit the ability of Pink1 grab parkin (Click here for more on this).
And we see this in flies.

Flies. Source: TheConservation
Flies (or drosophila) are a regular feature in biological research. Given their short life cycle, they can be used to quickly determine the necessity and function of particular genes. Yes, they are slightly different to us, but quite often the same biological principles apply.
Take Pink1 for example.
When scientists mutate the Pink1 gene in flies, it leads to the loss of flight muscles and male sterility. These effects both appear to be due to the kind of mitochondrial issues we were discussing above. One really amazing fact is that the human version of Pink1 can actually rescue the flies that have their Pink1 gene mutated. This is remarkable because across evolution genes begin to differ slightly resulting in some major differences by the time you get to humans. The fact that Pink1 is similar between both flies and humans shows that it has been relatively well conserved (functionally at least).
And given that we see similarities in the Pink1 gene and function between flies and humans, then perhaps we can apply what we see in flies to humans with regards to treatments.
Which brings us (finally!) to the research paper we wanted to look at today:

Title: Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease<
Authors: Lehmann S, Loh SH, Martins LM.
Journal: Biol Open. 2016 Dec 23. pii: bio.022186.
PMID: 28011627 (this article is OPEN ACCESS if you would like to read it)
In this study, the researchers analysed Pink1 flies and found alterations in the activity of an enzyme called nicotinamide adenine dinucleotide (or NAD+). NAD+ is one of the major targets for the anti-aging crowd and there is some very interesting research being done on it (Click here for a good review on this). NAD+ is a coenzyme found in all living cells. A coenzyme functions by carrying electrons from one molecule to another (Click here for a nice animation that will explain this better). The researchers found that Pink1 mutant flies have decreased levels of NAD+.
The researchers were curious if a diet supplemented with the NAD+ would rescue the mitochondrial defects seen in the Pink1 mutant fly. Specifically, they fed the flies a diet high in the NAD+ precursor nicotinamide (being a precursor means that nicotinamide can be made into NAD+ once inside a cell). They found that not only did nicotinamide rescue the mitochondrial problems in the flies, but it also protected neurons from degeneration.
So why is the title of this post talking about Niacin and not nicotinamide?
Niacin (also known as vitamin B3 or nicotinic acid) – like nicotinamide – is also a precursor of NAD+. And in their discussion of the study, the researchers noted that a high level of dietary niacin has been associated with a reduced risk of developing Parkinson’s disease (Click here and here for more on this).
The researchers were quick to point out that while a high Niacin diet may be beneficial, it could not be considered a cure in anyway for people with Parkinson’s disease because although it may be able to slow the cell death it would not be able to replace the cells that have already been lost.
So what does it all mean?
Hang on a second. We’re not finished yet.
Numerous media outlets have made a big fuss about the Niacin diet angle to this research, and they have ignored another really interesting finding:
In their study the researchers mutated another gene in the Pink1 flies which also resulted in improved mitochondrial function and neuroprotection. That gene was Poly (ADP-ribose) polymerase (or PARP). Parp is an enzyme involved in DNA repair and cell division. It is produced in very high levels in many types of cancer and medication that inhibit or block Parp are being tested in the clinic as therapies in those cancers.
Interestingly, blocking Parp has been previously shown to be beneficial for cell survival in models of Parkinson’s disease (Click here and here for more information on this). So in addition to changing to a high niacin diet, it would be interesting to follow up this results as well.
Particularly for people with the Pink1 mutation.
And this is where the results of this study are particularly interesting: they may relate specifically to a small population within the Parkinson’s community – those with Pink1 mutations. It would be interesting to begin discussing and designing clinical studies that focus particularly on people in this population (similar to the Ambroxol study – click here for our post on this).
So what does it all mean? (again)
The results of the present study demonstrate two means by which people with a particular genetic mutation could be treated for Parkinson’s disease. Obviously further research is required, but the idea that we are approaching an age in Parkinson’s disease research where treatments could be personalised is very appealing. It will be interesting to see where all of this goes.
EDITOR’S NOTE: If nothing we have written here makes any sense, then maybe this video will help:
The banner for today’s post was sourced from Wallpapersinhq



