A couple of very interesting studies were published a week ago that help us to better understand how we move. They are particularly important with respects to Parkinson’s disease.
The parts of the brain involved in movement
Movement is largely controlled by the activity in a specific collection of brain regions, collectively known as the ‘Basal ganglia‘.

The location of the basal ganglia structures (blue) in the human brain. Source: iKnowledge
The basal ganglia receives signals from the overlying cortex, processes that information before sending the signal on down the spinal cord to the muscles that are going to perform the movement.
There is also another important participant in the regulation of movement: the thalamus.

A brainscan illustrating the location of the thalamus in the human brain. Source: Wikipedia
The thalamus is a structure deep inside the brain that acts like the central control unit of the brain. Everything coming into the brain from the spinal cord, passes through the thalamus. And everything leaving the brain, passes through the thalamus. It is aware of most everything that is going on and it plays an important role in the regulation of movement.
The direct/indirect pathways
The processing of movement in the basal ganglia involves a direct pathway and an indirect pathway. In simple terms, the direct pathway encourages movement, while the indirect pathway does the opposite (inhibits it). The two pathways work together like a carefully choreographed symphony.
The motor features of Parkinson’s disease (slowness of movement and resting tremor) are associated with a breakdown in the processing of those two pathways, which results in a stronger signal coming from the indirect pathway – thus inhibiting/slowing movement.
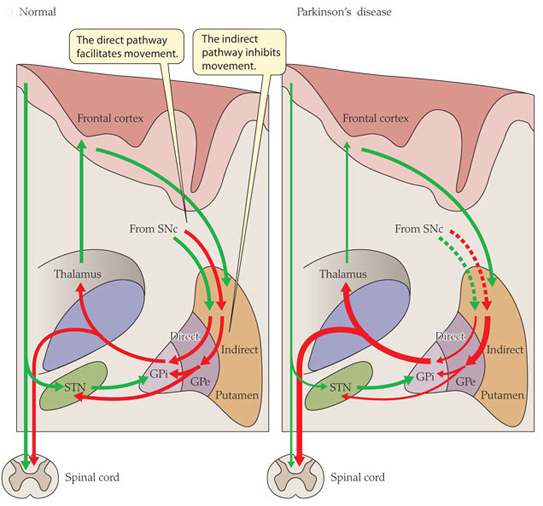
Excitatory signals (green) and inhibitory signals (red) in the basal ganglia, in both a normal brain and one with Parkinson’s disease. Source: Animal Physiology 3rd Edition
Both the direct and indirect pathways finish in the thalamus, but their effects on the thalamus are very different. The direct pathway leaves the thalamus excited and active, while the indirect pathway causes the thalamus to be inhibited.
The thalamus will receive signals from the two pathways and then decide – based on those signals – whether to send an excitatory or inhibitory message to the cortex, telling it what to do (‘get excited and movement’ or ‘don’t get excited and don’t move’, respectively).
Where does dopamine come into the picture?
In Parkinson’s disease, the cells in the brain that produce the chemical dopamine are lost. These cells reside in a structure called the substantia nigra (or SNc in the figure above). What effect does this cell loss have on the direct and indirect pathways? Under normal circumstances the dopamine neurons excite the direct pathway and inhibit the indirect pathway.
In Parkinson’s disease the loss of dopamine neurons results in increased activity in the indirect pathway. As a result, the thalamus is kept inhibited. With the thalamus subdued, the overlying motor cortex has trouble getting excited, and thus the motor system is unable to work properly.
So what was published last week?
Two papers.
Both from the same lab (Well done!)
One in the prestigious scientific journal, Cell and the other in her sister journal, Neuron:

Title: Cell-Type-Specific Control of Brainstem Locomotor Circuits by Basal Ganglia.
Authors: Roseberry TK, Lee AM, Lalive AL, Wilbrecht L, Bonci A, Kreitzer AC.
Journal: Cell, 2016 Jan 28;164(3):526-537.
PMID: 26824660
The researchers in this study discovered signal sent from the basal ganglia that selectively activates a group of neurons an area of the brainstem called the ‘mesencephalic locomotor region’. Some of the neurons in this area release a chemical called glutamate. Glutamate is a neurotransmitter that excites the cells it comes into contact with. The researchers who conducted this study found that these glutamate-releasing cells in the mesencephalic locomotor region are responsible for initiating movement.

The researchers used a new technique called ‘optogenetics’ that allows light to activate or inhibit specific cells in the brain. By using this technique on the cells in the direct (dMSN in the figure above) or indirect pathways (iMSN) of the basal ganglia, the researchers were able to control the glutamate-releasing neurons in the mesencephalic locomotor region of mice -initiating or inhibiting their movement, respectively.
The researchers then took the study one step further and used the optogenetics approach directly on the glutamate-releasing neurons in the mesencephalic locomotor region, and they were able to control the initiation of movement in the mice irrespective of the signal being generated by the direct or indirect pathways. That is to say, when the glutamate-releasing neurons in the mesencephalic locomotor region were activated, the mouse would move even when the basal ganglia was sending an inhibitory signal.
So what does it all mean?
While some of the findings of the study were already known, the researchers here have elegantly linked the workings of the basal ganglia and the mesencephalic locomotor region, helping us to better understand the neurological functioning of movement. Deep brain stimulation of the mesencephalic locomotor region has already been attempted and it has demonstrated mixed results in people with Parkinson’s disease (it does appear to help with regards to reducing falls – click here and here for more on this).
It will be interesting to follow the research resulting from this current study.

Title: Pathway-specific remodeling of thalamostriatal synapses in Parkinsonian mice
Authors: Parker PRL, Lalive AL, Kreitzer AC.
Journal: Neuron, 2016
PMID: 26833136
In the second study, the researchers (the same folks who gave us the first paper!) found that the basal ganglia is biased towards the direct pathway. The signal coming from the neurons involved in the direct pathway are stronger than those in the indirect pathway. When dopamine is removed however (as in the case of Parkinson’s disease), the system swings in the opposite direction and becomes biased toward the indirect pathway – the neurons in the direct pathway begin to produce a weaker signal than their counters in the indirect pathway which increase the strength of their signal.
Given that both pathways influence the activity of the thalamus, the researchers next turned their attention to that structure. Again using the ‘optogenics‘ (light-activation) technique, the investigators reduced the inhibitory signal coming from the thalamus and were able to reversibly correct the motor impairs observed in the mice with Parkinson’s-like features.
What does this mean for Parkinson’s disease?
This study turns our attention away from what is happening in the basal ganglia and focuses it on the thalamus, which has not receive the same amount of attention with regards to Parkinson’s disease.
There is a lot already known about changes in the thalamus in Parkinson’s disease (click here for more on this), and deep brain stimulation of structures in neighbouring regions is a regular therapy for Parkinson’s disease (targeting the subthalamic nuclei). But this new paper further breaks down the circuitry of movement for us and offers novel directions for future therapeutic approaches for Parkinson’s disease.
We can be sure that a lot of Parkinson’s disease research is now going to focus on the thalamus.