
Please excuse our use of UK slang in the title of this post, but a group of Australian researchers have recently discovered something really interesting about Parkinson’s disease.
And being a patriotic kiwi, it takes something REALLY interesting for me to even acknowledge that other South Pacific nation. This new finding, however, could be big.
In today’s post, we will review new research dealing with a protein called SOD1, and discuss what it could mean for the Parkinson’s community.

The number of dark pigmented dopamine cells in the substantia nigra are reduced in the Parkinson’s disease brain (right). Source: Adaptd from Memorangapp
Every Parkinson’s-associated website and every Parkinson’s disease researchers will tell you exactly the same thing when describing the two cardinal features in the brain of a person who died with Parkinson’s disease:
- The loss of certain types of cells (such as the dopamine producing cells of the substantia nigra region of the brain – see the image above)
- The clustering (or aggregation) of a protein called Alpha synuclein in tightly packed, circular deposits, called Lewy bodies (see image below).

A Lewy body inside a cell. Source: Adapted from Neuropathology-web
The clustered alpha synuclein protein, however, is not limited to just the Lewy bodies. In the affected areas of the brain, aggregated alpha synuclein can be seen in the branches of cells – see the image below where alpha synuclein has been stained brown on a section of brain from a person with Parkinson’s disease.

Examples of Lewy neurites (indicated by arrows). Source: Wikimedia
Now, one of the problems with our understanding of Parkinson’s disease is disparity between the widespread presence of clustered alpha synuclein and very selective pattern of cell loss. Alpha synuclein aggregation can be seen distributed widely around the affected areas of the brain, but the cell loss will be limited to specific populations of cells.
If the disease is killing a particular population of cells, why is alpha synuclein clustering so wide spread?
So why is there a difference?
We don’t know.
It could be that the cells that die have a lower threshold for alpha synuclein toxicity (we discussed this is a previous post – click here?).
But this question regarding the difference between these two features has left many researchers wondering if there may be some other protein or agent that is actually killing off the cells and then disappearing quickly, leaving poor old alpha synuclein looking rather guilty.

Poor little Mr “A Synuclein” got the blame, but his older brother actually did it! Source: Youtube
And this is a very serious discussion point.
This year of 2017 represents the 200th anniversary of James Parkinson’s first description of Parkinson’s disease, but it also represents the 20th anniversary since the association between alpha synuclein and PD was first established. We have produced almost 7,000 research reports on the topic of alpha synuclein and PD during that time, and we currently have ongoing clinical trials targetting alpha synuclein.
But what if our basic premise – that alpha synuclein is the bad guy – is actually wrong?
Is there any evidence to suggest this?
We are just speculating here, but yes there is.
For example, in a study of 904 brains, alpha synuclein deposits were observed in 11.3% of the brains (or 106 cases), but of those cases only 32 had been diagnosed with a neurodegenerative disorder (Click here to read more on this). The remaining 74 cases had demonstrated none of the clinical features of Parkinson’s disease.
So what else could be causing the cell death?
Well, this week some scientists from sunny Sydney (Australia) reported a protein that could fit the bill.

Sydney. Source: Vagabond
The interesting part of their finding is that the protein is also associated with another neurodegenerative condition: Amyotrophic lateral sclerosis.
Remind me again, what is Amyotrophic lateral sclerosis?
Parkinson’s disease and Amyotrophic lateral sclerosis (ALS) are the second and third most common adult-onset neurodegenerative conditions (respectively) after Alzheimer’s disease. We recently discussed ALS in a previous post (Click here to read that post).
ALS, also known as Lou Gehrig’s disease and motor neuron disease, is a neurodegenerative condition in which the neurons that control voluntary muscle movement die. The condition affects 2 people in every 100,000 each year, and those individuals have an average survival time of two to four years.
You may have heard of ALS due to it’s association with the internet ‘Ice bucket challenge‘ craze that went viral in 2014-15.

The Ice bucket challenge. Source: Forbes
What is the protein associated with ALS?
In 1993, scientists discovered that mutations in the gene called SOD1 were associated with familial forms of ALS (Click here to read more about this). We now know that mutations in the SOD1 gene are associated with around 20% of familial cases of ALS and 5% of sporadic ALS.
The SOD1 gene produces an enzyme called Cu-Zn superoxide dismutase.
This enzyme is a very powerful antioxidant that protects the body from damage caused by toxic free radical generated in the mitochondria.

SOD1 protein structure. Source: Wikipedia
One important note here regarding ALS: the genetic mutations in the SOD1 gene do not cause ALS by affecting SOD1’s antioxidant properties (Click here to read more about this). Rather, researchers believe that the cell death seen in SOD1-associated forms of ALS is the consequences of some kind of toxic effect caused by the mutant protein.
So what did the Aussie researchers find about SOD1 in Parkinson’s disease?
This week, the Aussie researchers published this research report:

Title: Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated withneuronal loss in Parkinson’s disease brain.
Authors: Trist BG, Davies KM, Cottam V, Genoud S, Ortega R, Roudeau S, Carmona A, De Silva K, Wasinger V, Lewis SJG, Sachdev P, Smith B, Troakes C, Vance C, Shaw C, Al-Sarraj S, Ball HJ, Halliday GM, Hare DJ, Double KL.
Journal: Acta Neuropathol. 2017 May 19. doi: 10.1007/s00401-017-1726-6.
PMID: 28527045
Given that oxidative stress is a major feature of Parkinson’s disease, the Aussie researchers wanted to investigate the role of the anti-oxidant enzyme, SOD1 in this condition. And what they found surprised them.
Heck, it surprised us!
Two areas affected by Parkinson’s disease – the substantia nigra (where the dopamine neurons reside; SNc in the image below) and the locus coeruleus (an area in the brain stem that is involved with physiological responses to stress; LC in the image below) – exhibited little or no SOD1 protein in the control brains.
But in the Parkinsonian brains, there was a great deal of SOD1 protein (see image below).

SO1 staining in PD brain and Control brains. Source: Springer
In the image above, you can see yellowish-brown stained patches in both the PD and control images. This a chemical called neuromelanin and it can be used to identify the dopamine-producing cells in the SNc and LC. The grey staining in the PD images (top) are cells that contain SOD1. Note the lack of SOD1 (grey staining) in the control images (bottom).
Approximately 90% of Lewy bodies in the Parkinson’s affected brains contained SOD1 protein. The investigators did report that the levels of SOD1 protein varied between Lewy bodies. But the clustered (or ‘aggregated’) SOD1 protein was not just present with alpha synuclein, often it was found by itself in the degenerating regions.
The researchers occasional saw SOD1 aggregation in regions of age-matched control brains, and they concluded that a very low level of SOD1 must be inherent to the normal ageing process.
But the density of SOD1 clustering was (on average) 8x higher in the SNc and 4x higher in the LC in the Parkinsonian brain compared to age-matched controls. In addition, the SOD1 clustering was significantly greater in these regions than all of the non-degenerating regions of the same Parkinson’s disease brains.
The investigators concluded that these data suggest an association between SOD1 aggregation and neuronal loss in Parkinson’s disease. Importantly, the presence of SOD1 aggregations “closely reflected the regional pattern of neuronal loss”.
They also demonstrated that the SOD1 protein in the Parkinsonian brain was not folded correctly, a similar characteristic to alpha synuclein. A protein must fold properly to be able to do it’s assigned jobs. By not folding into the correct configuration, the SOD1 protein could not do it’s various functions – and the investigators observed a 66% reduction in SOD1 specific activity in the SNc of the Parkinson’s disease brains.
Interestingly, when the researchers looked at the SNc and LC of brains from people with ALS, they identified SOD1 aggregates matching the SOD1 clusters they had seen in these regions of the Parkinson’s disease brain.
Is this the first time SOD1 has been associated with Parkinson’s disease?
No, but it is the first major analysis of postmortem Parkinsonian brains. SOD1 protein in Lewy bodies has been reported before:

Title: Cu/Zn superoxide dismutase-like immunoreactivity is present in Lewy bodies from Parkinson disease: a light and electron microscopic immunocytochemical study
Authors: Nishiyama K, Murayama S, Shimizu J, Ohya Y, Kwak S, Asayama K, Kanazawa I.
Journal: Acta Neuropathol. 1995;89(6):471-4.
PMID: 7676802
The investigators behind this study reported SOD1 protein was present in Lewy bodies, in the substantia nigra and locus coeruleus of brains from five people with Parkinson’s disease. Interestingly, they showed that SOD1 is present in the periphery of the Lewy body, similar to alpha synuclein. Both of these protein are present on the outside of the Lewy body, as opposed to another Parkinson’s associated protein, Ubiquitin, which is mainly present in the centre (or the core) of Lewy bodies (see image below).
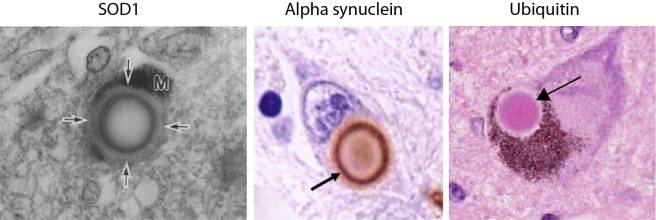
A more recent study also demonstrated SOD1 protein in the Parkinsonian brain, including direct interaction between SOD1 and alpha synuclein:

Title: α-synuclein interacts with SOD1 and promotes its oligomerization
Authors: Helferich AM, Ruf WP, Grozdanov V, Freischmidt A, Feiler MS, Zondler L, Ludolph AC, McLean PJ, Weishaupt JH, Danzer KM.
Journal: Mol Neurodegener. 2015 Dec 8;10:66.
PMID: 26643113 (This article is OPEN ACCESS if you would like to read it)
These researchers found that alpha synuclein and SOD1 interact directly, and they noted that Parkinson’s disease related mutations in alpha synuclein (A30P, A53T) and ALS associated mutation in SOD1 (G85R, G93A) modify the binding of the two proteins to each other. They also reported that alpha synuclein accelerates SOD1 aggregation in cell culture. This same group of researchers published another research report last year in which they noted that aggregated alpha synuclein increases SOD1 clustering in a mouse model of ALS (Click here for more on this).
We should add that alpha synuclein aggregations in ALS are actually quite common (click here and here to read more on this).
Are there any genetic mutations in the SOD1 gene that are associated with Parkinson’s disease?
Two studies have addressed this question:

Title: Sequence of the superoxide dismutase 1 (SOD 1) gene in familial Parkinson’s disease.
Authors: Bandmann O, Davis MB, Marsden CD, Harding AE.
Journal: J Neurol Neurosurg Psychiatry. 1995 Jul;59(1):90-1.
PMID: 7608718 (This article is OPEN ACCESS if you would like to read it)
And then in 2001, a second analysis:

Title: Genetic polymorphisms of superoxide dismutase in Parkinson’s disease.
Authors: Farin FM, Hitosis Y, Hallagan SE, Kushleika J, Woods JS, Janssen PS, Smith-Weller T, Franklin GM, Swanson PD, Checkoway H.
Journal: Mov Disord. 2001 Jul;16(4):705-7.
PMID: 11481695
Both studies found no genetic variations in the SOD1 gene that were more frequent in the Parkinson’s affected community than the general population. So, no, there are no SOD1 genetic mutations that are associated with Parkinson’s disease.
Are there any treatments targeting SOD1 that could be tested in Parkinson’s disease?
Great question. Yes there are. And they have already been tested in models of PD:

Title: The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease.
Authors: Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, White AR, Crouch PJ, Lim S, Leong SL, Wilkins S, George J, Roberts BR, Pham CL, Liu X, Chiu FC, Shackleford DM, Powell AK, Masters CL, Bush AI, O’Keefe G, Culvenor JG, Cappai R, Cherny RA, Donnelly PS, Hill AF, Finkelstein DI, Barnham KJ.
Title: J Exp Med. 2012 Apr 9;209(4):837-54.
PMID: 22473957 (This article is OPEN ACCESS if you would like to read it)
CuII(atsm) is a drug that is currently under clinical investigation as a brain imaging agent for detecting hypoxia (damage caused by lack of oxygen – Click here to read more about this).
The researchers conducting this study, however, were interested in this compound for other reasons: CuII(atsm) is also a highly effective scavenger of a chemical called ONOO, which can be very toxic. CuII(atsm) not only inhibits this toxicity, but it also blocks the clustering of alpha synuclein. And given that CuII(atsm) is capable of crossing the blood–brain barrier, these investigators wanted to assess the drug for its ability to rescue model of Parkinson’s disease.
And guess what? It did!
And not just in one model of Parkinson’s disease, but FOUR!
The investigators even waited three days after giving the neurotoxins to the mice before giving the CuII(atsm) drug, and it still demonstrated neuroprotection. It also improved the behavioural features of these models of Parkinson’s disease.
Is CuII(atsm) being tested for anything else in Clinical trials?
Yes, there is a clinical trial ongoing for ALS in Australia.
The Phase I study, being run by Collaborative Medicinal Development Pty Limited, is a dose escalating study of Cu(II)ATSM to determine if this drug is safe for use in ALS (Click here for more on this study).

Cu(II)ATSM is an orally administered drug that inhibits the activity of misfolded SOD1 protein. It has been shown to paradoxically increase mutant SOD1 protein in a mouse model of ALS, but it also provides neuroprotection and improves the outcome for these mice (Click here to read more on this).
If this trial is successful, it would be interesting to test this drug on a cohort of people with Parkinson’s disease. Determining which subgroup of the Parkinson’s affected community would most benefit from this treatment is still to be determined. There is some evidence published last year that suggests people with genetic mutations in the Parkinson’s associated gene PARK2 could benefit from the approach (Click here to read more on this). More research, however, is needed in this area.
So what does it all mean?
Right, so summing up, a group of Australian researchers have reported that the ALS associated protein SOD1 is closely associated with the cell death that we observe in the brains of people with Parkinson’s disease.
They suggest that this could highlight a common mechanisms of toxic SOD1 aggregation in both Parkinson’s disease and ALS. Individuals within the Parkinson’s affected community do not appear to have any genetic mutations in the SOD1 gene, which makes this finding is very interesting.
What remains to be determined is whether SOD1 aggregation is a “primary pathological event”, or if it is secondary to some other disease causing agent. We are also waiting to see if a clinical trial targeting SOD1 in ALS is successful. If it is, there may be good reasons for targeting SOD1 as a novel treatment for Parkinson’s disease.
The banner for today’s post was sourced from Pinterest

 Title: Miro1 Marks Parkinson’s Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson’s Models.
Title: Miro1 Marks Parkinson’s Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson’s Models.


















{kind=link}