![]()
|
Gaucher disease is a genetic disorder caused by the reduced activity of an enzyme, glucocerebrosidase. This enzyme is produced by a region of DNA (or a gene) called GBA – the same GBA gene associated with a particular form of Parkinson’s. Recently, a Danish company has been testing a new drug that could benefit people with Gaucher disease. It is only natural to ask the question: Could this drug also benefit GBA-associated Parkinson’s? In today’s post, we will discuss what Gaucher disease is, how this experimental drug works, and why it would be interesting to test it in Parkinson’s. |

Will Shakespeare. Source: Ppolskieradio
The title of this post is a play on words from one of the many famous lines of William Shakespeare’s play, Hamlet.
The original line – delivered by Marcellus (a Danish army sentinel) after the ghost of the dead king appears – reads: If the authorities knew about the problems and chose not to prevent them, then clearly something is rotten in the state of Denmark.
(Act 1, Scene 4)
The title of this post, however, is: Something is interesting in the state of Denmark
This slight change was made because certain Danish authorities know about the problem and they are trying to prevent it. The ‘authorities’ in this situation are some research scientists at a biotech company in Denmark, called Orphazyme.
And the problem is Parkinson’s?
No, the problem is Gaucher disease.
Huh? What is Gaucher disease?
Gaucher disease (pronounced “go-shay”) is a rare inherited genetic disorder characterised by the build up in cells of a fatty chemical called glucocerebroside. Because the body cannot break down this chemical, swollen fat-laden cells build up in certain areas of the body, such as the spleen, liver and bone marrow. These cells are referred to as ‘Gaucher cells’.

Swollen Gaucher cells (circled in red). Source: Imagebank
The disorder results from the deficiency of the enzyme glucocerebrosidase, which usually breaks down glucocerebroside. The incidence of Gaucher disease is about one in every 40,000 live births (Source), and the condition manifests itself in several way, from reduced bone density to swollen liver and spleen:

The signs of Gaucher disease. Source: Rare2aware
When you say Gaucher disease is a rare genetic disorder, what does that mean?
Gaucher disease is caused by genetic mutations in a gene called ‘GBA’.
What is ‘GBA’?
GBA is a gene – a section of DNA that provides the instructions for making a particular protein. In the case of the GBA gene, the protein is an enzyme, called Glucocerebrosidase.
What does Glucocerebrosidase do?
Glucocerebrosidase (also known as GCase) helps with the digestion and recycling of glucocerebrosides inside cells. The enzyme is located and active inside ‘lysosomes‘.
What are Lysosomes?
Lysosomes are small bags of degradative enzymes – like glucocerebrosidase – that can be found inside of cells.
On a continual regular basis, small parts of the external layer of the cell membrane is brought inside the cell. This is a process called endocytosis. It occurs when the cell consumes resources from the outside world in order to find what it needs to function and survive. As a section of cell membrane is brought into the cell, it forms a vesicle (which is a term used to refer to small spherical bags of stuff inside cells). Given the process by which that vesicles was formed, it is referred to as an endosome (sometimes it is also called a vacuole).

Source: Socratic
Once the endosome is inside the cell and detached from the rest of the membrane, it will bind to another vesicle which is called a lysosome. And as I mentioned above, lysosome is a small bag that is full of digestive enzymes, which help to break down the contents of the endosome.

How lysosomes work. Source: Prezi
The lysosome will fuse with the endosome/vacuole and the enzymes from the lysosome will mix with the material in the vacuole and digest it (or it break down into more manageable components).
This enzymatic process works in a very similar fashion to the commercial products that you use for washing your clothes.

Enzymatic degradation. Source: Samvirke
The reagents that you put into the washing machine with your clothes contain a multitude of enzymes, which help to break down the dirty, bacteria, flakes of skin, etc that cling to your clothes. Each enzyme breaks down a particular protein, fat or such like. And this is very similar to the collection of enzymes in the lysosome. All of them are needed to break down all of the contents of the endosome.
And if one of those enzymes – such as glucocerebrosidase – is faulty (due to a mutation in the DNA providing the instructions for its construction), then the enzymatic process is disrupted, which could result in the build up of un-degraded material over time.
And since glucocerebrosidase is faulty in Gaucher disease, the condition is referred to as a lysosomal storage condition.
What happens to the cells in Gaucher disease?
Macrophage are one type of cell that is particularly affected in Gaucher disease. They are a type of blood cell that is responsible for detecting, engulfing and destroying dangerous pathogens and apoptotic cells. Below is a schematic of a macrophage, consuming orange pathogens (left), digesting them, and releasing the waste (on the right):

A schematic of a macrophage. Source: Meducator
Macrophage literally travel the body, swallowing anything that they don’t like the look of. In order to break down everything it swallows, a macrophage must have a full complement of digestive enzymes. But – as you can see in the image below – without glucocerebrosidase, the macrophage has trouble digesting fatty chemicals like glucocerebrosides and the lysosomes start to accumulate in the cell, causing the cell to swell up.
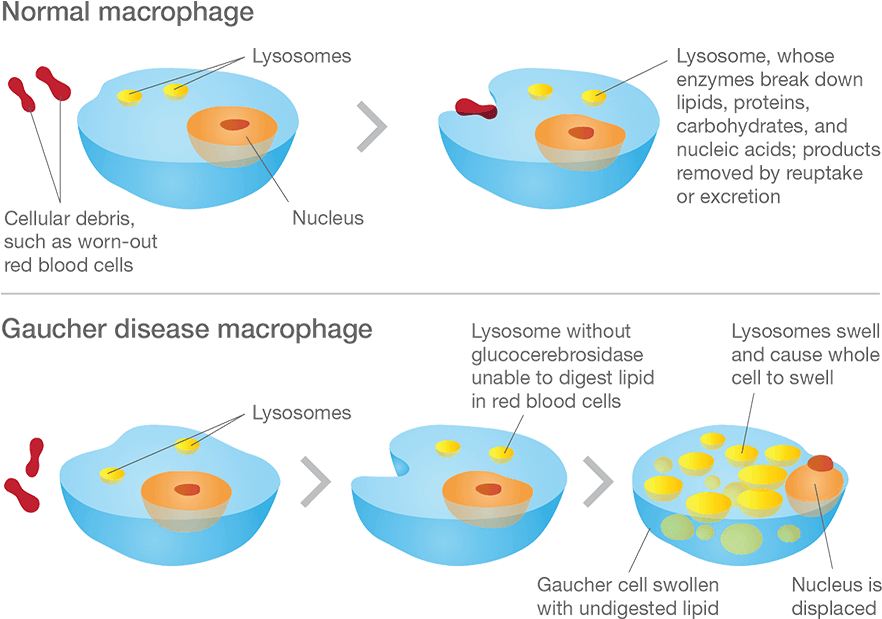
Gaucher disease. Source: Gaucherawareness
Are there different types of Gaucher disease?
There are three types of Gaucher disease, and they are all caused by genetic mutations:
- Type I – (also called the “non-neuropathic” type) this is the most common; having said that it mainly occurs in Ashkenazi Jews (x100 more than the general population). The median age at diagnosis is 28 years of age, and life expectancy is only mildly decreased. As the “non-neuropathic” label suggests, there are no neurological symptoms.
- Type II – is characterised by neurological problems in small children. The glucocerebrosidase enzyme is barely present in the lysosomes. Prognosis is poor (death before the age of three).
- Type III – (the Swedish variety) occurs in people from the Norrbotten region in Sweden. This group develops the disease somewhat later, but most most do not survive their 30th birthday.
There are three subtypes of Gaucher disease, which are characterised by the presence or absence of neurological issues, and by the rate of disease progression and severity:
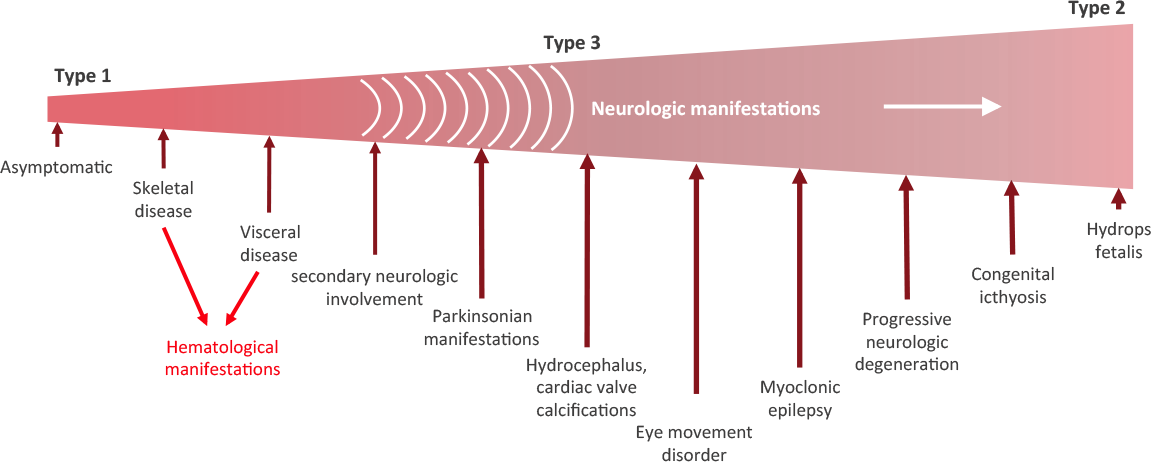
Stages of Gaucher disease. Source: Gaucherawareness
You said Gaucher disease is inherited. So if one of my parents has Gaucher disease will it affect me?
As I mentioned above Gaucher disease is an inherited genetic disorder, caused by mutations in the GBA gene. Humans normally have two copies of the GBA gene. If one copy of the GBA gene is faulty due to a genetic mutation, the person will not develop Gaucher’s disease, because the one remaining functional gene will be able to produce enough of the glucocerebrosidase enzyme.
Gaucher disease is considered an autosomal recessive disorder. This means that two copies of an abnormal gene must be present in the individual in order for the disease to develop. A person with just one faulty gene will not get sick, but they will be a carrier. To develop Gaucher disease, you need to have two genetic mutations in the GCase gene – one from your mother and one from your father.
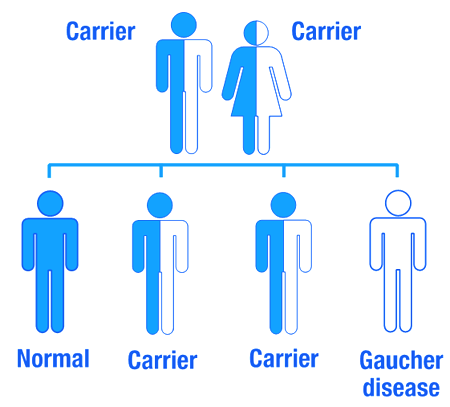
An autosomal recessive disorder. Source: Myhealthyfeeling
So even if one of your parents has been diagnosed with Gaucher disease, you will not necessarily develop it if the other parent does not have a mutation in their GBA gene.
Ok. All of this is interesting, but how is Gaucher disease associated with Parkinson’s?
In the 1990s, physicians began to notice patients with both Gaucher and Parkinson’s. An example of this was a report published in 1996 that described six people with Gaucher disease who also exhibited an early-onset, severe form of Parkinson’s with cognitive decline:

Title: Occurrence of Parkinson’s syndrome in type I Gaucher disease.
Authors: Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A.
Journal: QJM. 1996 Sep;89(9):691-4.
PMID: 8917744 (This article is OPEN ACCESS if you would like to read it)
In this study, the Israeli researchers report on 6 people with Type I Gaucher disease (which up until that point had not been considered neuronopathic). All six of the subjects also exhibited the hallmark of a rather severe form of Parkinson’s, which made its appearance in the 4th to 6th decade of life and displayed an aggressive progression and was largely unresponsive to conventional anti-Parkinson therapy (such as L-dopa).
These initial reports were followed by many additional studies which eventually started pointing towards the GBA gene as the likely risk factor for this form of Parkinson’s, including this study:

Title: Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews.
Authors: Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R.
Journal: N Engl J Med. 2004 Nov 4;351(19):1972-7.
PMID: 15525722 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators examined 99 individuals with idiopathic Parkinson’s from an Ashkenazi Jewish background. Thirty-one of them (31.3%) had one or two mutations in their GBA gene. And of all the individuals with Parkinson’s, the subjects who were carriers of GBA mutations were younger than those who were not carriers (mean age at onset being 60 years vs. 64 years).
And this result is similar to what has been seen in larger follow up studies (Click here for an example).
It is now believed that approximately 5%–8% of people with Parkinson’s have a genetic mutation in the GBA gene (Click here and here to read more about this). According to the Michael J Fox foundation webpage on GBA “up to 10 percent of people with PD in the United States carry” a genetic variation in the GBA gene.
GBA is a very large gene and there are numerous genetic variants spread across its length. The most common mutations are located in positions N370S and L444P. These mutations cause a reduction in the enzymatic activity of the glucocerebrosidase enzyme.
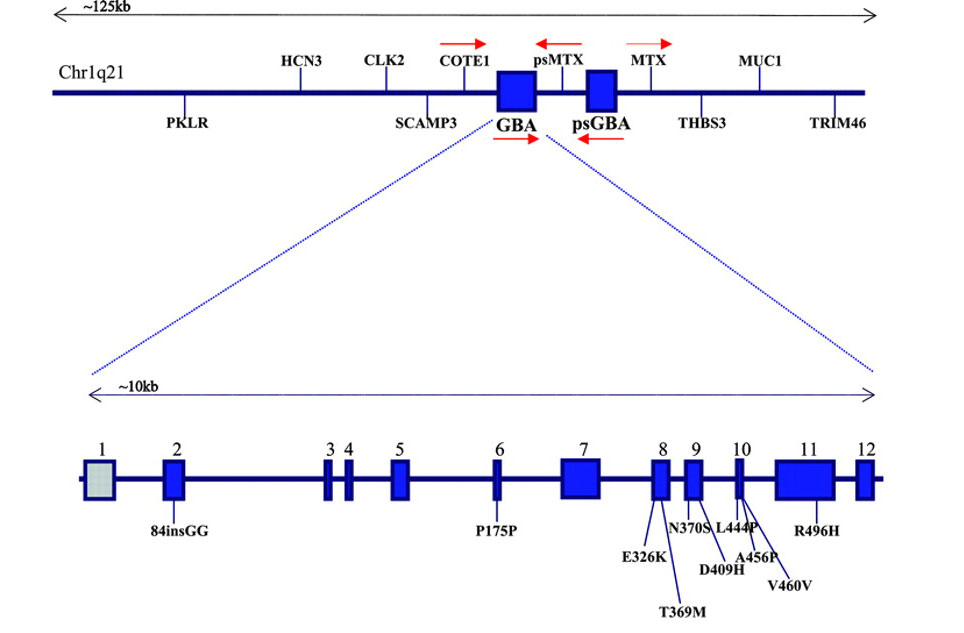
The GBA gene and mutations associated with Parkinson’s. Source: Neurology
And as I mentioned above generally, people with GBA-associated Parkinson’s will exhibit more severe symptoms than people without a GBA mutation, and this has recently been confirmed in a large clinical observation study:

Title: Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study.
Authors: Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, Bajaj N, Barker RA, Burn DJ, Foltynie T, Hardy J, Wood NW, Ben-Shlomo Y, Williams NW, Grosset DG, Morris HR; PRoBaND clinical consortium.
Journal: J Neurol Neurosurg Psychiatry. 2018 [Epub ahead of print]
PMID: 29378790 (This article is OPEN ACCESS if you would like to read it)
In this multi-research centre study, the investigators studied 1893 people with Parkinson’s. Of these 48 (2.5%) had one copy of a known GBA mutation, 117 (6.2%) had a non-GBA genetic variant that had previously been associated with Parkinson’s (such as mutations in genes like LRRK2 or Alpha Synuclein), and 28 (1.5%) carried genetic variants of unknown significance in the GBA gene. As I mentioned above, onte of the most common GBA mutations associated with Parkinson’s is called ‘L444P‘ and this was also the most common GBA mutation observed in this study.
Individuals with Parkinson’s associated GBA mutations were on average diagnosed 5 years earlier compared with non-carriers. They were also more likely to have postural instability and gait difficulties compared with non-carriers. In addition, they had more progressive forms of Parkinson’s (as determined by more advanced Hoehn and Yahr staging – after adjustment for age – compared with non-carriers). Curiously, no differences was observed in cognitive function between GBA mutation carriers and non-carriers. Cognitive impairment/dementia have been reported in other studies at a later stages of the condition (Click here to read more about this). This observation led the researchers to conclude that “this offers an important window of opportunity for potential disease-modifying therapy that may protect against the development of dementia”.
This is really bad, right?
This aggressive pattern of disease progression in GBA-associated Parkinson’s is not always the case. Plus, 2-3% of the general population will have a Parkinson’s associated GBA mutation in their DNA, but never present any features of Parkinson’s. And then there are cases of identical twins who both have a GBA mutations, but only one of them has developed Parkinson’s (Click here to read more about this).
Thus, the genetics of Parkinson’s is still very complex, and just because a person has a GBA mutation, it does not necessarily mean that they will go on to develop Parkinson’s or necessarily present the aggressive form of the condition. More research is required to increase our understanding of this situation.
In addition, there are already numerous clinical trials ongoing for drugs targeting GBA-associated Parkinson’s (CLick here to learn more about that). And novel potential Gaucher/Parkinson’s drugs are being identified all the time.
Which brings us to the research report this post will be reviewing. Last week, this manuscript appeared on the BioRxiv website:

Title: Arimoclomol as a potential therapy for neuronopathic Gaucher Disease
Authors: Fog-Tonnesen CK, Zago P, Malini E, Solanko LM, Peruzzo P, Bornaes C, Magnoni R, Petersen NHT, Bembi B, Dardis A, and Kirkegaard T
Journal: bioRxiv preprint first posted online Mar. 15, 2018.
PMID: N/A (this article is OPEN ACCESS if you would like to read it)
In this preprint manuscript (meaning that this research has not yet been through the peer-review process), the researchers were interested in assessing the impact of Arimoclomol on cells collected from people with Gaucher Disease.
What is Arimoclomol?
Arimoclomol is an experimental, orally administered drug that was originally developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. This compound is designed to stimulate a protein repair pathway by activating compounds called “molecular chaperones”.
What are molecular chaperones?
Molecular chaperones are simply proteins that assist in the folding (or unfolding) and the assembly (or disassembly) of other proteins. They are like the make-up artists and wardrobe departments back stage at the theatre, making sure the actors are correctly dressed and ready for the big stage.

Source: Biosocialmethods
A newly-formed amino acid chain can fold itself into a protein, but the assistance of molecular chaperones helps ensure that everything proceeds smoothly and quickly, regardless of whatever other events might be occurring in the cell.
Has much research been conducted with Arimoclomol?
Quite a lot actually.
But the real excitement started in 2004, when researchers noticed something interesting:

Title: Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice.
Authors: Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L.
Journal: Nat Med. 2004 Apr;10(4):402-5. Epub 2004 Mar 21.
PMID: 15034571
In this study, the researchers gave arimoclomol to a mouse model of ALS.
What is ALS?
Amyotrophic lateral sclerosis (or ALS; also known as motor neuron disease), is a neurodegenerative condition in which the neurons that control voluntary muscle movement die. The condition affects 2 people in every 100,000 each year, and those individuals have an average survival time of two to four years.

ALS in a nutshell. Source: Walkforals
ALS is a progressive neurodegenerative disease, characterised by the death of motor neurons in the motor cortex of the brain, the brain stem, and the spinal cord. This results in muscle weakness and atrophy throughout the body, causing the affected individual to ultimately lose the ability to initiate and control all voluntary movement.

Motor neurons. Source: Alsfoundation
ALS is also known as Lou Gehrig’s disease after the New York Yankees baseball player who developed the condition:

Lou Gehrig. Source: NBC
In 1969, Henry Louis “Lou” Gehrig was voted the greatest first baseman of all time by the Baseball Writers’ Association. He played 17 seasons with the New York Yankees, having signed with his hometown team in 1923.
For 56 years, he held the record for the most consecutive games played (2,130), and he was only prevented from continuing that streak when he voluntarily took himself out of the team lineup on the 2nd May, 1939, after his ability to play became hampered by the disease that now often bears his name. A little more than a month later he retired, and a little less than two years later he passed away.
Another famous individual affected by ALS was the theoretical physicist, Prof Stephen Hawking (who passed away last week):

Prof Stephen Hawking. Source: BBC
He was diagnosed with in a very rare early-onset, slow-progressing form of ALS in 1963 (at age 21) that has gradually left him wheel chair bound.
There is no cure for ALS and a medication called riluzole only extends life by two to three months.
And what is SOD1?
It is estimated that 5 to 10% of all ALS cases are due to an inherited genetic mutation. One of those mutations occurs in the superoxide dismutase 1 (SOD1) gene. This gene provides that instructions for a protein that is important for the detoxification of motor neurons (we have previously discussed SOD1 on this blog – Click here to read that post). In SOD1-associated ALS, a mutant version of the SOD1 protein is produced in the motor neurons, and the mutant protein does not do its job properly.
In ALS research, scientists have generated mice that produce the mutant version of SOD1 protein and it results in the death of motoneurons. This cell loss may be due to the availability of molecular chaperones, so researchers have been testing to determine if increasing molecular chaperone activity in ALS mice could rescue the model.
When researchers treated SOD1-ALS mice with arimoclomol from a young age (35 days of age), they found a marked improvement in muscle function and motoneuron survival in the later stages of the disease, which resulted in a 22% increase in lifespan. They also saw a significant improvement in the mice when they delayed the starting the arimoclomol treatment until the symptoms started to appear at 70 days of age. In the graph below, you can see the lifespan of untreated SOD1-ALS mice (Black line) compared to those treated with arimoclomol from 35 (red line) or 70 (green line) days of age.

Source: Nature
In a follow up study, the investigators found that treatment after 90 days has no significant effect on lifespan. They also noticed that arimoclomol reduced the protein aggregation that is observed in these SOD1-ALS mice (Click here to read more about that follow up study).
Other research groups have also tested arimoclomol, and some of that work has been conducted in other models neurodegenerative conditions. For example, this study:

Title: Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses.
Authors: Kirkegaard T, Gray J, Priestman DA, Wallom KL, Atkins J, Olsen OD, Klein A, Drndarski S, Petersen NH, Ingemann L, Smith DA, Morris L, Bornæs C, Jørgensen SH, Williams I, Hinsby A, Arenz C, Begley D, Jäättelä M, Platt FM.
Journal: Sci Transl Med. 2016 Sep 7;8(355):355ra118.
PMID: 27605553
In this study, the researchers tested arimoclomol in both cell culture and two independent mouse models of Niemann-Pick disease type C (or NPC). It is associated with genetic mutations in NPC1 and NPC2 genes. NPC is a lysosomal storage disease – similar to Gaucher disease. But rather than a build up of glucocerebrosides (as is the case with Gaucher disease), in NPC there is an accumulation of cholesterol and other lipids due to a reduction in the lysosomal enzyme called acid sphingomyelinase.
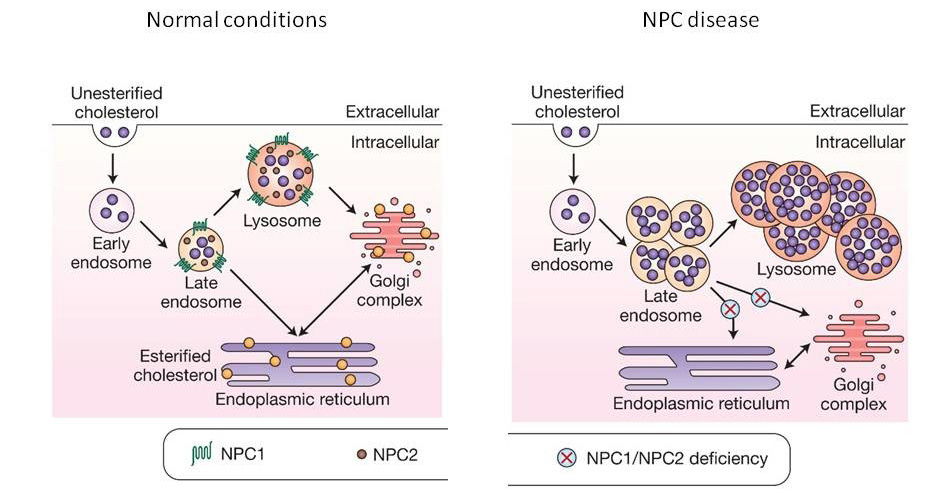
Niemann-Pick disease type C. Source: irb
In this study, the researchers found that arimoclomol treatment of cell cultures of cells from people with NPC could reduce the burden of cholesterol accumulation in these cells. They also found beneficial effects in mouse models of NPC, including the activation of molecular chaperones in the brain.
Has Arimoclomol been tested in clinical trials?
Yes it has.
In 2008, the results of a phase I clinical trial of arimoclomol in amyotrophic lateral sclerosis reported that the drug was found to be safe and well tolerated at dosages up to 300 mg/day (Click here to read more about this).
Then in February this year, researchers from Florida, Boston & Sweden published this clinical study report:

Title: Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS.
Authors: Benatar M, Wuu J, Andersen PM, Atassi N, David W, Cudkowicz M, Schoenfeld D.
Journal: Neurology. 2018 Feb 13;90(7):e565-e574.
PMID: 29367439 (this article is OPEN ACCESS if you would like to read it)
In this study, the researchers conducted a double-blind, placebo-controlled Phase 2/3 trial of arimoclomol on 38 individuals with rapidly progressive SOD1 amyotrophic lateral sclerosis (Click here to read more about this trial). Of the participants recruited to the study, 19 were given placebo and 17 received arimoclomol. The researchers found that arimoclomol was safe and well-tolerated at a dosage of 200 mg per day (for up to 12 months) – there were very few adverse events (including side effects). Importantly, the participants on arimoclomol declined more slowly as determined by the Revised ALS Functional Rating Scale (ALSFRS-R) and the forced expiratory volume in 6 seconds (FEV6) test. As you can see in the image below, at 12 months, 34% of arimoclomol-treated were alive, compared to <21% of placebo-treated participants.

Source: NCBI
The investigators were quick to point out, however, that this study was “not powered for therapeutic effect”, meaning that they didn’t have enough participants to determine if the effect was statistically significant. But they are very keen to further investigate the use of arimoclomol in ALS.
A larger follow-up study is planned to start in 2018.
But before everyone gets too excited, there have also been studies which have questioned the utility of arimoclomol in certain conditions. For example:

Title: Targeting protein homeostasis in sporadic inclusion body myositis.
Authors: Ahmed M, Machado PM, Miller A, Spicer C, Herbelin L, He J, Noel J, Wang Y, McVey AL, Pasnoor M, Gallagher P, Statland J, Lu CH, Kalmar B, Brady S, Sethi H, Samandouras G, Parton M, Holton JL, Weston A, Collinson L, Taylor JP, Schiavo G, Hanna MG, Barohn RJ, Dimachkie MM, Greensmith L.
Journal: Sci Transl Med. 2016 Mar 23;8(331):331ra41.
PMID: 27009270 (this article is OPEN ACCESS if you would like to read it)
Sporadic inclusion body myositis is an acquired progressive muscle disorder. It is the most common severe myopathies (conditions in which the muscle fibers do not function properly) in people over age 50 years of age. The degenerative aspect of the condition is characterised by the appearance of holes in the vacuoles of muscle cells, and the accumulation of abnormal proteins within the cells.
In this study, the researchers found that treatment with arimoclomol reduced disease pathology and improved muscle function in animal models of sporadic inclusion body myositis, but when they then shifted to a randomised, double-blind, placebo-controlled, proof-of-concept clinical trial of arimoclomol in people with sporadic inclusion body myositis, the drug had no impact on the condition. It was safe and well tolerated, but “there was no statistically significant difference in the secondary outcome measures favouring Arimoclomol” (primary outcome was safety; secondary outcome was efficacy).
So what were the results of the BioRxiv study?
Ah, yeah. I almost forgot (man, what a long and convoluted post!).
So, last week, this manuscript appeared on the BioRxiv website:
Title: Arimoclomol as a potential therapy for neuronopathic Gaucher Disease
Authors: Fog-Tonnesen CK, Zago P, Malini E, Solanko LM, Peruzzo P, Bornaes C, Magnoni R, Petersen NHT, Bembi B, Dardis A, and Kirkegaard T
Journal: bioRxiv preprint first posted online Mar. 15, 2018.
PMID: N/A (this article is OPEN ACCESS if you would like to read it)
In this study, researchers from Ospedali Riuniti Udine (Italy) and a Danish biotech company Orphazyme A/S conducted experiments testing arimoclomol on cells collected from people with Gaucher disease. They found that arimoclomol induced molecular chaperones and enhanced the folding, maturation, activity and the correct cellular localisation of mutated GCase protein. The results were replicated across cells from several forms of Gaucher disease (including the common L444P and N370S mutations).
In the graphs below you can see the level of GCase activity in cells treated with different concentrations of arimoclomol for 1 – 5 days (left hand graph), but the researchers found that long-term (4 weeks) arimoclomol treatment on Gaucher cells at very low doses could significantly increase the levels of GCase activity (right hand graph) – back to levels equal to that of normal/wild type (WT) control cells that were not treated with arimoclomol.

Source: BioRxiv
One can only wonder if similar beneficial results would be observed in cell culture models of GBA-associated Parkinson’s.
Has arimoclomol ever been tested in models of Parkinson’s?
Not that I’m aware of.
But given the association between Gaucher disease and GBA-associated Parkinson’s it would certainly be very interesting for someone to have a look.
I’m just saying.
Orphazyme A/S?
Are you reading this??
What does it all mean?
A biotech company in Denmark has put some interesting research into the public domain that suggests one of their drugs, called arimoclomol, could provide benefits to people suffering from Gaucher disease. Gaucher’s is a rare genetic disease caused by specific mutations in the Parkinson’s-associated gene, GBA. Given that these two conditions share a similar genetic risk factor, it could be interesting to test this drug in the context of GBA-associated Parkinson’s.
Pre-clinical data will be required. And that research would also be able to determine if arimoclomol has any impact on other Parkinson’s associated proteins such as alpha synuclein. But given that numerous clinical trials have already been conducted with arimoclomol, taking it to the clinic for GBA-associated Parkinson’s would be a relatively straightforward task.
I think it’s an idea that may be worth further investigation. Orphazyme A/S? You hearing me???
Addendum – 28th September, 2018
Today Orphazyme reported the results of their Phase II/III clinical trial of Arimoclomol in Niemann-Pick disease Type C (NPC; a rare lysosomal condition which we discussed in the post above).
They found that Arimoclomol was well-tolerated over a 12 month period in 50 patients with NPC. And the top-line results show a treatment benefit of arimoclomol over placebo on the primary endpoint 5-domain NPC Clinical Severity Scale (NPC-CSS) (p-value 0.07 – not statistically significant, but terribly close!). To read more about thsese results – Click here.
Orphazyme is currently discussing with the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) what the best path is towards making arimoclomol available to those suffering from NPC.
Positive stuff.
But again, isn’t it time for some research in GBA-associated Parkinson’s???
EDITOR’S NOTE: Orphazyme is publicly traded company. That said, the material presented on this page should under no circumstances be considered financial advice. Any actions taken by the reader based on reading this material is the sole responsibility of the reader. Orphazyme has not requested that this material be produced, nor has the author had any contact with this company or any associated parties. This post has been produced for educational purposes only.
The banner for today’s post was sourced from Orphazyme

I haven’t even given this its due yet, but I know a lady who blogs for the Huff: Elaine Benton http://elainebenton.blogspot.co.uk/
She suffers both Gaucher’s and Parkinson’s. It’s been hellacious for her. I’ll be back when I’ve done you justice and read the article, Simon!!
LikeLike
Okay – read and re-read, Simon. I’ll have to do several reads just to get it right! Have you approached Orphazyme with this? Beautiful science!
LikeLike
Hi Lisa,
I haven’t approached them (except tongue in cheek on social media). It is a bit outside of my area of expertise, and I am assuming that they will already be looking at it. But you know what they say about assumptions. Hopefully by bringing arimoclomol to more peoples attention, someone will pick up the ball and run with it. Interesting drug.
Kind regards,
Simon
LikeLiked by 1 person