
|
This is one of those posts (read: rants) where I want to put an idea out into the ether for someone to chew on. It starts with a very simple question: Why is ‘the drug’ the focus of a clinical trial? If our goal is to find beneficial therapies for people with Parkinson’s, then the way we currently clinically test drugs is utterly nonsensical. And if we do not change our “we’ve always done it this way” mindset, then we are simply going to repeat the mistakes of the past. Others are changing, so why aren’t we? In today’s post, we will consider one possible alternative approach. |

I hope you know who Grace Hopper is – if not, click here. Source: Mentalfloss
Why is ‘the drug‘ the focus of a clinical trial?
The way we clinically test drugs makes absolutely no sense when you actually stop and think about it.
Other medical disciplines (such as oncology) have woken up to this fact, and it is time for the field of Parkinson’s research to do this same.
Let me explain:
The easiest way to test a novel treatment for a medical condition is to simply give it to your population of interest and see what happens.
For example, we have a drug (let’s call it ‘Curetide’ – à la a previous rant) and we would like to test it on a group of people with a particular medical condition (for the rest of this post we will use the example of Parkinson’s). So, we take a group of 20-30 people with Parkinson’s and we give them Curetide to take over a certain period of time, and we conduct assessments on each of them to see if the drug is having any effect. At the end of our study, we pool all the data together and look to see if Curetide had any beneficial effect on their Parkinson’s features.
This approach begins with the rather ridiculous assumption that everyone affected by Parkinson’s is the same, and that they are affected by the condition in the same way. If you have ever walked into a Parkinson’s support group meeting, you will know that reality is somewhat different from the theoretically ideal situation.

What if the drug only works for the red person?
Not only are people different, but the way their bodies process drugs varies significantly as well. Thus, in the schematic above, our Parkinson’s affected population should not be viewed as a equal population, but rather in different shades of different colours.
Now, to counter this problem of heterogeneity (individual differences) within our affected population of interest, our current clinical testing system takes the very prudent step of comparing our drug of interest with a neutral control (or placebo) treatment. A placebo is a treatment that looks identical to the drug of interest (in this case ‘Curetide’ – trademark pending by the way!) in every respect, except that it has no biological effect – in drug testing the placebo pill contains an ‘inactive substance’. This placebo is given to a control group in parallel to the treatment group, and it is usually given in a ‘blinded’ manner (meaning that the participants in the study are unaware of which treatment they are receiving – they do not know if they are being given the drug being tested or the placebo).
In an ideal world, this situation would look something like this:

And the only difference between the two groups would be the drug being tested – everything else would be exactly the same. The potential therapeutic benefit of the drug is being compared to the effect of a neutral placebo treatment between two identical groups.
But again, reality is not very accommodating:

As the image above suggests, both groups in any clinical trial will made up of a mixed bunch of individuals.
Now, some very sharp readers will point out that the two groups are still identical. Thanks to the variability between individual participants within each group, the two groups will balance out – they will shift to some kind of “Parkinson’s average” – and thus, the two groups can be considered equal. In theory, this would return us confidently to a situation where the drug is being compared to the placebo (between two almost equal groups).
There may be some truth to this.
And lest we forget, “we have always done it this way”.
But this is where the whole system begins to fail the patients.
Our current system requires a drug like Curetide to have a huge effect on the ‘treatment group’. A lot of people within the treatment group need to exhibit positive outcomes from taking Curetide. If the drug does not have a big enough beneficial effect on enough people in the treatment group (when compared to the control group), it will not succeed in passing through the various stages of the clinical trial process. It will therefore fail, and Curetide will be sent to the ever-growing scrapheap of failed drugs.
Under our current system, we basically have a filtering system for blockbuster drugs.
This approach may be considered great for the large pharmaceutical companies, whose business models are designed around developing drugs that can be sold to large proportions of the population.
But the whole system fails to appreciate an troublesome fact: the fact that we are all unique.
Let’s reconsider the situation from above once more:
Two identical groups. One experimental drug (Curetide) being tested. And let’s keep the two groups on the drug (or placebo) for 12 months and then see what effect the drug has had.
What could possibly go wrong?
Hypothetical question for you: what happens if I tell you that unbeknownst to us, Curetide only works in the ‘red’ individuals in the image above?
Will the drug be successful in the current clinical trial system? Will Curetide have a big enough impact on the results of the treatment group for it to pass the assessment process?
Unlikely.
The average person in the treatment group will exhibit no difference when compared to the control group, and this will result in Curetide failing to demonstrate any effect in the context of the whole trial… even though it may have been very effective for one particular person. In fact, that one ‘responder’ in the treatment group would probably be considered an outlier, and the drug will be quickly shipped off to that ever-growing scrapheap of failed drugs I mentioned above.

Not looking good. Source: Pixabay
In every clinical trial of an experimental treatment, there will be responders and non-responders to the treatment.
As the label on the can suggests, ‘responders’ are individuals who exhibit a positive (or – in really bad situations – a negative) response to the treatment, while a ‘non-responder’ presents a response to the treatment that is no better than any of the members of the control group. The distribution of these groups can vary from treatment to treatment – some drugs will have a lot of responders, while others will have very few – but there are almost always responders and non-responders in the treatment group of a clinical trial.
The difference between responders and non-responders is based on our individual biology.
For example, in 2016, researchers discovered that there are hundreds of people living in Norway who are completely unresponsive to opioid-based pain killers (such as morphine) due to a genetic variation in their DNA (Source). In an opiate-based pain killer drug trial before 2016, these individuals would probably have been the non-responders in a clinical trial treatment group, potentially lessening the chances of success for the experimental pain killer.
So basically, our current system is a filter for treatments that have the most responders.
And while this may at first appear to be a useful approach for the pharmaceutical industry in their efforts to identify blockbuster drugs, in truth billions of dollars of investor money are being wasted on ‘failed’ trials where there were not enough positive responders in the treatment group. It is wasteful approach that does not serve the affected community nor society as a whole. And critically, it is reducing our chances of overall success by limiting the number of potentially useful drugs.
There is the very real possibility that effective drugs (for certain individuals) are being lost or missed out on because our current system is flawed.

A hopeless loss. Source: Matt Lamers on Unsplash
If our goal is to truly find beneficial therapies for people with Parkinson’s, then the system needs to change.
We have to shift our approach from filtering for drugs that treat the maximum number of responders, to simply filtering patients.
Let me explain:
Take a group of people diagnosed with Parkinson’s. Give them an experimental drug (for example, Curetide) and then monitor them over a period of time. Maybe no one responds to that drug – fine, but after a period of time we move everyone to the next drug and repeat the process. Those who respond to that second drug stay on the second drug. Those who do not respond (the non-responders) get shifted to the next drug. And this process would continue thus:

One critical part of such a system would be feedback.
The characteristics that define a person who responds to a particular drug would be collected and fed back to the clinicians, thus allowing for better determination of who might respond to that drug in the future. New participants and new drugs could be added to such a study design as it proceeds, adding to the pool of information being collected and fed back to help better assign individuals.
Another critical part of such a system would be continuous measures of assessment which would be assessed continuously and acted on. That is to say, if it is apparent that a drug is having no impact, the individual would be shifted to another drug (as opposed to waiting till the end of a classical clinical trial to determine if a drug has actually had any impact). These assessments would require us to learn as much as we can about each person enrolled in such a study, from their genetic risk factors and biochemistry through to their environment.
And the adaptive nature of this system would allow for different combinations of drugs over time. For example, after a certain period of time the responders to drug no.2 in the image above, could be given drug no.3 to see if any additive benefits could be gained. If they respond well to the combination of drug no. 2+3, then they would stay on that regime. If they didn’t exhibit any additional benefits, they could be shifted to a combination of drug no.2+4 (as indicated in the image below):

Such an additive approach would work well for a condition like Parkinson’s where ‘a cure’ would require a multi-component treatment approach will be likely (Click here to read more about this).
Overall, when compared to the classical clinical trial, this new style of clinical trial is considered adaptive. An adaptive clinical trial “evaluates a medical device or treatment by observing participant outcomes (and possibly other measures, such as side-effects) on a prescribed schedule, and modifying parameters of the trial protocol in accord with those observations” (Source). To read more about adaptive clinical trials – click here for a good review.
And a lot of clinical trials are now incorporating aspects of the adaptive trial idea. In fact, in 2013 it was calculated that approximately 20% of Phase III trials utilise some elements of adaptive design (Source).
The most popular type of adaptive clinical trial is referred to as a multi-arm, multi-stage (MAMS) platform trial. These compare multiple drugs at the same time – stopping an drugs that fail to demonstrate benefits, while continuing with those that do. But thus far most of these trials have been conducted on the group level (rather than individual patients), and thus are still under that influence of the “maximum number of responders” effect we mentioned above. In addition, many of them have not included cross-over components (where participants are shifted to new drugs).
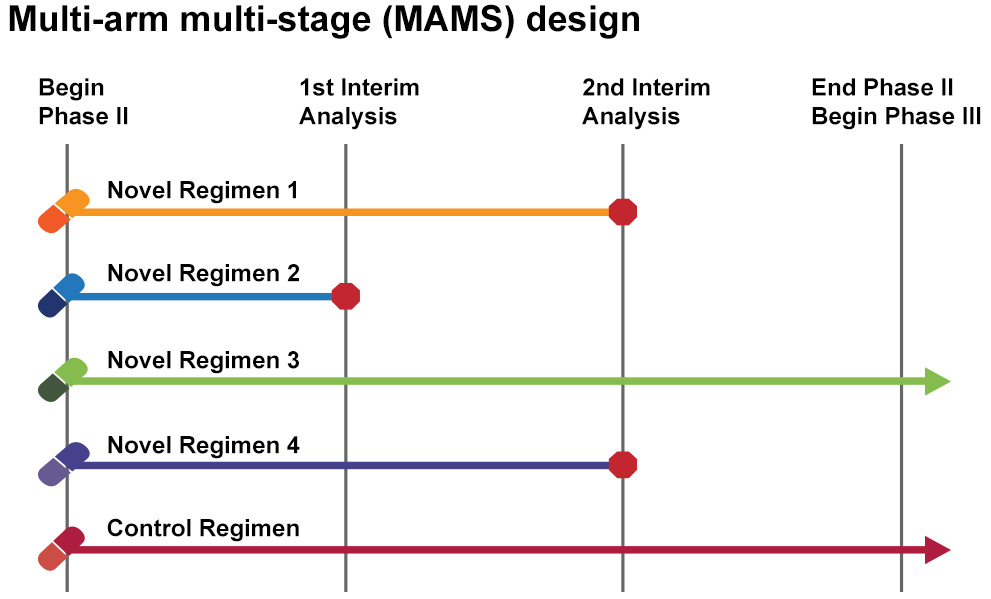
Multi-arm, multi-stage (MAMS) platform trial. Source: Eupati
One of the best examples of an adaptive clinical trial are the I-SPY studies – which are focused on cancer.

Source: ispytrials
I-SPY (“Investigation of Serial studies to Predict Your therapeutic response with imaging and molecular analysis”) is an adaptive trial design that has enabled two experimental breast cancer drugs to deliver promising results after just six months of clinical testing (those drugs being Veliparib and Neratinib – Click here to read more about this). The value of the I-SPY trials, however, go well beyond the clinical results, as they have given proof of principle validation to the concept, resulting in a host of additional adaptive clinical trial projects for various medical conditions, such as Prostate cancer (the Stampede trials), Multiple Sclerosis (the MS-Smart trial) and Alzheimer’s (the EP-AD consortium).
Which begs the obvious question: why are Parkinson’s clinical trials lagging behind this trend? Where is the adaptive trial for Parkinson’s?
And rather than simply jumping on the band wagon, why shouldn’t an adaptive Parkinson’s clinical trial attempt something innovative and radical – raising the bar further for adaptive trials and providing the template for something new?

New opportunity. Photo by Tanja Heffner on Unsplash
There are several good reasons for the lag.
- Cost: Adaptive clinical trials to date have been very expensive to set up and run. But if we compare that to the cost of all the failed clinical trials of the past, perhaps the cost is not so great.
- Definitions: Given the variety we see in individuals with the condition, it is tempting to ask ‘what exactly is Parkinson’s?’ While cancers have very specific biopsy-based definitions of certain tumors, Parkinson’s is only just coming up with some basic groupings of subgroups based on genetic risk factors. Hopefully, however, characterising responders to a particular drug in an adaptive clinical trial might highlight novel groupings.
- Assessment: My haters (loveable as I am, I do have a few) will say that we are not there yet with biomarkers and tools of assessment, particularly if we are going to measure benefits in individual patients. But this only brings into question the methods we use in our current clinical trials (if we can’t measure individuals accurately, how do we measure groups of individuals accurately?). In truth, however, I am inclined to agree on this matter in the case of Parkinson’s. But we have to start somewhere, right?
There are also other confounding issues regarding this type of study:
- Adherence: How does an investigator make sure an individual participant sticks to the treatment regime? If the patient hears about some amazing new herbal remedy (in some dark corner of the interweb) or reads a newspaper headline about some new re-purposed drug for Parkinson’s, what is to stop them from trying it in a desperate effort to slow the condition? Such an action would potentially mess up their trial results.
- Choosing an intermediate outcome measures: How and when do the investigators track individuals over time and decide to shift participants to or from a particular drug? This comes back to the issue of assessment mentioned above (Click here for a technical read on this topic)
- Lifestyle: How can investigators account for life style factors like exercise when assessing/measuring individuals?
You can probably tell that the patient filtering idea proposed here is not perfect in the real world setting of the research clinic.
And there are certainly aspects of it that are completely impractical.
I do not profess to know everything or to have any of the answers, and I certainly do not care if this post makes me the subject of mockery or ridicule with my research colleagues. But what I would like to see is constructive discussion (involving the Parkinson’s affected community) regarding the current state of our clinical trials. And critically, the sharing of some thoughts/ideas regarding innovation in this important area.
In particular, I would like to see a discussion that questions the dogma and “the way we’ve always done it” mindset that surrounds our current approach to clinical trials.
* * * * * * * * * * * * * * * * * *
Hopefully this post is food for thought for someone.
The biggest human temptation is to settle for too little – Thomas Merton
The banner for today’s post was sourced from Searchengineland

Excellent and interesting post. Love those who think outside the box.
LikeLike
Hi Chrissy,
Glad you liked the post. And it’s not outside the box thinking, it’s re-thinking the box completely. Knocking out a wall and renovating. Re-doing the whole Feng shui of the box. Adding a mezzanine floor, and putting wheels on it.
Kind regards,
Simon
LikeLike
We need a leader for our group to advance these ideas
Sent from my iPhone
>
LikeLike
Hi Scott,
Are you volunteering?
Simon
LikeLike
Since you have some of the answers…how does gene mapping relate? Would that make this all more doable? Is this gene idea five years away?
I did take an herb for another condition that worked very well. When nih did their big study it was found ineffective c.f. to placebo. I guess that is your point. It was actually effective. My researcher at nih doctor was convinced too.
We all want a cure. Some folks want fame and fortune too. Your idea would still allow for fortune wouldn’t it? When 8 of 10 respond well to drug number 2, jackpot!
Anyway, just trying yo get my head around your idea. Sounds like the way to go. Thanks
LikeLike
Hi Dkdc,
Thanks for the comment. The gene mapping could be one way, but I would rather filter participants and find out who responds to a drug before then determining what characteristics those responders share (maybe it’s a particular genetic variant). The current system of taking mixed groups of PwPs and trying to find new biomarkers ultimately results in potentially useful biomarkers for subsets of PwPs being drowned out by the average of the whole PwP group (same story as the current approach to drug testing).
Do you still take the herb?
Kind regards,
Simon
LikeLike
Simon,
your idea of adaptive patient-centric trails is already reality for many of us – those who DO try the latest internet-hyped nutritional supplement, magic herbal elixir, lifestyle tweak… We just have an n=1 and no clinician to take objective measures of outcomes. Such subjective personal trails are of course of no use to anyone but the patient – but if I start taking Curetide and feel after two weeks that I’m better than when I wasn’t, does that subjectivity invalidate the usefulness of Curetide? Obviously not.
What if 100 others like me reported that Curetide made them “better” than they were? 1,000? As you know, such “crowd-sourced” treatment evaluation is the concept behind CliniCrowd (what is the update on Mannitol ?).
I agree that the the challenge to bringing objective clinical evaluation to this scenario lies in the lack of readily available assessment tools (DATScan doesn’t qualify, spinal taps for a-syn probably don’t either) – enter Simon’s PD breathalyzer (how’s that coming along by the way?) – something like that would be game changer.
On your concerns regarding “Confounding issues” – 1. Adherence – those exact same concerns and issues prevail in current “drug-centric” trials too (it would not be a problem specific to the new adaptive trial modality). 2. Choosing outcomes – in the patient-centric mode, the outcome is relative to the individual i.e. does the patient do better than they were doing before? If the result is equivocal, then we could subtract the treatment and observe to see if they start to revert. I don’t see a problem with this. 3. Lifestyle – as in #2 above, the outcome measures for the treatment are relative to where the individual is starting from.
LikeLiked by 1 person
Hi Alex,
Thanks for your insightful comment. You are right that many in the community are already doing their own N=1 type of ‘studies’. But for the medical world, it would be useful if such information was being collected and analysed, hence the need to these adaptive style trials.
The “breathalyzer” results are coming along – 170 PD subjects and 22 controls enrolled (anyone wondering what we’re talking about click here: https://scienceofparkinsons.com/breath-analysis-study/). We have just conducted some biased analysis (selecting variables like years since diagnosis and comparing chemicals in the breath) and the AI machine is now going to start the harder (but potentially more useful) unbiased approach (matching groups based on chemicals and looking for common clinical variables). I will be presenting the biased results at the Parkinson’s UK Gretschen Amphlett memorial lecture in Cambridge, UK on the 17th April which will be streamed live over the web (Eeek! https://www.youtube.com/watch?v=KxoKOWvCwDU ). But I can tell you that Prof Haick and his team (the inventors of the breath analysis technology) are now conducting a clinical study of their first cell phone prototype – each time you make a phone call the device analyses your breath! So all I can suggest is to watch this space regarding that tech.
The issue with “Confounding issues” #2. Choosing outcomes is the ‘when’ and ‘how’ to measure issue – 2 weeks after starting the treatment or 6 months? And how depends on the anticipated outcome (something mitochondrial for example could be easy to measure via a blood analysis test, but for drugs with more complicated mechanisms of action it may be more difficult). And with #3., if the participant changes exercise regime or diet (etc) after shifting drugs and has a beneficial effect, which was the influencer? These N=1 studies will need to be more carefully controlled than the current large group clinical trials. But then if we are collecting more info from each participant (continuous data from smart pills and other gadgets) maybe it would be easier.
I contacted Clinicrowd several months back for an update post, but I got no response. I’ll try again and let you know.
Kind regards,
Simon
LikeLike
This is interesting! In breast cancer they have the brca and other genes that help clarify treatment responses. But in PD it seems all over the map. Even folks with lrrk2 mutations have can have such different responses. Maybe this kind of approach would help bring clarity about the genetics in PD. Even if they stick with the same style of research it would seem smart to get genetic info on everyone in the treatment group so they could check for correlations.
LikeLike
Hi Diana,
Glad you found it interesting.
Genetics sounds very sexy, but it is still such a messy business. BRCA mutations increase risk, but they are not definitive (only Huntington’s is definitive – with regards to neuro conditions). But you are right that filtering patients with drugs could possibly help to point towards some clarity in the genetics. And you are also right that it would be a case of collecting as much information as you possibly can about participants in the study (genetics included), and finding the similarities between responders.
Kind regards,
Simon
LikeLike
Simon,
Excellent. Your article covers a vast range of issues.
Do we run clinical trials to do good science? To get regulatory approval? To get a premium price? To improve the lot of the patient?
Regarding n=1 trials, the key concept to get across is “If it works for me, it works for me regardless of whether it works for you.” In fact, it is a bit more complicated than that. This is because our assessment of whether the therapy “works” for me is never completely accurate, and so we can learn, but not too much, from other people’s experiences. For instance, suppose I was taking X and it appeared to be working. How would I react if someone else who was also taking X died?
Regarding the treatment of the placebo effect in trials, we need to see the placebo effect as a friend, rather than as an enemy that adds to the complexity of testing. The effectiveness of a drug is influenced by many things: the consultation with a doctor, the packaging, the advertising etc., and not just the chemicals within it. This leads to “If it works for me, it works for me regardless of why.” Once again it is more complicated than that: I’d prefer a treatment which had firm foundations in both theory and practice. Given their power to do good, we could usefully manufacture placebos.
Nothing is purely therapeutic or placebo; even water would likely have a true therapeutic effect. So I would introduce a new term “therebo” ( from a potential therapy or placebo). We could even more usefully manufacture therebos. For instance, a special vitamin formulation for PD would be a therebo: it might have a true therapeutic effect or it might not.
John
LikeLike
Hi John,
Thanks for your comment – glad you liked the post. Lots of interesting ideas/themes here (“someone on X dying” – wow, I think that would mess with my head if I was on X). I like ‘therebo’ – I’m going to start using that (even though it is the same as placebo, as opposed to nocebo). Therebo has a better ring to it.
But an important question here is what do we mean by “it works for me” – specifically how do we determine what works? If we blindly give PwPs water and tell them it has miraculous preclinical results, perhaps we will see a placebo response, but will we see improvements on a DATscan?
Kind regards,
Simon
LikeLike
Hi Simon, this is a great build on your recent post, “The Dilemma of Success”. I agree completely that things need to change. We all know that Parkinson’s is a highly variable condition; from person to person, from day to day, from drug response to drug response. So why is there such inertia (rhetorical question)?
Here’s an extract from my comment on the previous post, which I think is relevant to this one
“Perhaps the worst-case scenario is a therapy that produces a statistically significant result; but one which is modest, and homogeneous amongst all PwP. It may then be obligatory in future trials, but will dial down the differential, meaning that future therapies would need to be particularly effective, and trial numbers large, to reach statistical significance. It would also disrupt the outcome of an adaptive trial.”
The trial designs you describe seem to depend on heterogeneous responses. Presumably if drug 2 produced such results, you’d then put them all onto drug 3?
Another issue, mentioned by John, is safety. The mindset of the regulators may still be one of numbers – prove it’s safe in a sufficiently large patient group. Would they wish to see safety data in the “efficacious” population?
This relates to the next point. It makes a lot of sense to do the MAMS or I-SPY design with existing drugs, where safety is established. It then becomes possible to test the principle of these designs for Parkinson’s primarily on efficacy.
Bearing in mind the rigidity of clinical trial protocols, in that you have to do what you say you’re going to do, why not have an end point as “to discover the percentage of responders to Curetide, where a responder is defined as………”
The next consideration is the purpose of the therapy. Is it for symptom relief? Slowing or stopping the progression? Replacing what has been lost?
Finally (for now, anyway…..), my impression is that current trials recruit people with a positive PD diagnosis. Perhaps we should be more discriminating about the stage of disease to be tested, whether that derives from prodromal symptoms, newly diagnosed, mid-stage or advanced. Every group has different challenges.
You’ve raised some very important issues here, ones which need to be discussed in the broader Parkinson’s community.
Kevin
LikeLike
Hi Kevin,
Thanks as always for the comment. You raise a lot of really important issues.
I probably should have mentioned in the post that this style of drug testing system would only work post phase I testing. We will always need phase I safety trials to determine that a compound is ok in man. And continuing with that theme, as I was writing this post I was only thinking of clinically available drugs which has been the focus of most MAMS thus far (as you say, we know a great deal about their safety track record). If curetide doesn’t work for anyone, shift the cohort to Exenatide. Non-responders to that will next shift to Nilotinib, then something else and so-on. And initially, slowing or stopping the progression would be the goal. I like the idea of stage specific testing, but again we would require stage specific biomarkers.
Ok, it’s late – more on all of this later.
Kind regards,
Simon
LikeLike
Simon
Assessing PD. There are various mobilephone applications and keyboard monitoring apps which are available now. and how is your research into breath going?
Keith
LikeLike
Hi Keith,
Thanks for your comment. You are right: Assessing PD is the big issue. We have loads of drugs we could test, but what are the read outs. I have to admit that I’m not a huge fan of the smart phone approach – A. a level of technical experience is required which my dad and some of his friends don’t really have; B. adherence (again), it would require a disciplined effort from the participant each day to keep inputting data. I much prefer the methods that require nothing from the individual, such as the smart pill approach (https://www.proteus.com/ – a pill that participants pop with their medication and it can measure various biological aspects as it passes through the system – no placebo effect there). The keyboard monitoring is a little more biased, but I like this idea and there is more research coming out on it (such as this research report published yesterday – https://www.jmir.org/2018/3/e89/) – though in the wake of Facebook/CambridgeAnalytica I am personally nervous about any keyboard recording apps. I would avoid online banking with such apps in the background 🙂
Regarding the breath analysis research – see my comment to Alex above.
Kind regards,
Simon
LikeLike
I am, inter alia, one of some 1500 cohort participants in the Oxford Parkinson’s Disease Centre‘s Monument Discovery Programme [OPDC]. In practical terms an annual assessment replicates the original interview (body fluids and party games), I receive offers to partake in research projects and get invited to interesting , often inspiring lectures
Some four years’ ago one such event entitled ‘Are there different types of Parkinson’s?’ explored the association of symptoms exhibited by the cohort .
Focussing on 5 classic symptoms of Parkinson’s, RBD (REM Sleep Disorder, Cognitive, Psychological Well Being, Tremor, non-tremor) the statistics associated people exhibiting these disorders into 5 clusters or ‘phenotypes’, imaginatively dubbed Groups 1 to 5 (To see the five groups, click here – https://content.iospress.com/articles/journal-of-parkinsons-disease/jpd140523#jpd-5-2-jpd140523-g002 and if you are interested in learning more about this research, click here to watch a video presentation about the results: https://www.youtube.com/watch?v=Keuy__i-d0A).
If nothing else it concisely illustrates this rather large cohort is indeed heterogeneous. People in Group 5, for example comprise 10.1% of the cohort and present far worse symptoms than all other Groups, whilst those of the first Group have a relatively milder prognosis (28.9%).
Whatever the reason for these differences, being aware would mean that a drug trial should be patient centric where trials are planned and performed in an adaptive manner rather than ‘one size fits all’.
For example a drug trial aimed at say Cognitive Disorders, would likely provide a different response for those in each particular group but differ most markedly between those in Groups 1 and 5
And a potentially effective drug may well have been saved from that scrap heap of failure.
If there are potentially significant benefits gained by adopting an adaptive, patient centric approach to drug trials, how may this be achieved?
If the OPDC were aware of this some five or more years’ ago maybe OPDC were encouraged by their cohort clustering to try out an adaptive approach for a drugs trial?? Has anyone asked them? If not, isn’t it perhaps the time to do so?
In a former not too distant life, I would expect any proposal for a major change to conducting business presented by way of a comprehensive ‘business case’.
I have no first-hand experience of drug trials, but moving to an adaptive model seems like major change to me. This proposal needs to be constructed and presented as a compelling business proposition that justifies moving from the tried and trusted way of doing things.
Who has to be persuaded? The major stakeholders, other than the ultimate stakeholders (those of us directly affected by Parkinson’s). These are the people who control the money, who fund the research, who expect a worthwhile ‘Return On Investment’, – that is, the Chief and Senior Executives, whatever their title, of the Parkinson’s charities, the government bodies and agencies , the corporate pharmaceutical collaborators.
It is this ‘investor cohort’ who must be convinced of the advantages of such a change. They do not necessarily all need be to convinced at once.
In recent years, other fields of clinical research have ventured into adaptive models of clinical trials, benefitting from being early adopters but running the risks that pioneering brings. By being a de facto follower Parkinson’s should avoid ‘proof of concept’ and similar risks that typify early adoption.
If there are examples of success in the areas of Prostate Cancer, Alzheimer’s, and MS, there must surely be some advocates, if not one or two zealots prepared to share the reasons for their success and help us build that compelling case. What CEO could fail to be interested in a drug trial that starts delivering useful information after only six months ?
The preferable suggestion may be to trial the approach with one supportive funding group, the Charities say. They would attempt to mandate the adaptive approach for all funding bids involving drug trialling and let the real benefits of this process be evidenced by the outcome .
‘That’s my suggestion, feel free to throw rocks at it.
LikeLike
Hi Roger,
Many thanks for your significant contribution to this discussion. It is greatly appreciated, and raises some important points.
The Oxford Parkinson’s Disease Centre (OPDC) Cohort is a fascinating study. An incredible treasure trove of data has been collected. And it is interesting to note that 18 minutes into the video link you have provided, Dr Michelle Hu makes the comment that if a drug works for group 5 (10% of the cohort), it would fail (I recommend readers to watch the video).
I will admit that I am aware of efforts being made to conduct a multi-arm, adaptive trial for Parkinson’s in the UK but they are still very much in the planning phase (I am not aware of anything in the US – but given how in vogue ‘adaptive’ is, there must be some efforts). Having said that I am not sure how much contribution is being made by members of the affected community to those UK efforts. And the purpose of the post was to make people aware of this style of trial and encourage feedback from the community.
In the UK, NICE (https://www.nice.org.uk/) and the MHRA (https://www.gov.uk/government/organisations/medicines-and-healthcare-products-regulatory-agency) are the regulators that need to be on board before any green light will be given. I suspect that funding would not be difficult to find (compared to a risky traditional clinical trial of something experimental), given the success of adaptive efforts in other conditions, but these adaptive trials are very expensive and thus it would need to be a huge chunk of funding.
The study would have to be multi-centre, but they would have to be big centres (taking on 40-50+ participants each). It would be pointless having small centres with 5 or 6 participants each. The cohort at each centre would have to be a priority for that centre, not a side project. And as I mentioned in the comment with Kevin, the drugs used in any first adaptive studies would best be clinically available drugs where we have a great deal of information regarding safety profiles (nothing too experimental). Yes, the issue then becomes getting Pharmas on board, but we are quickly reaching a point where enough off-patent drugs (Exenatide, Ambroxol, etc) are being proposed for re-purposing that this may not be too much of a problem. This would obviously reduce costs, allowing for a proof-of-principle trial to be run in which protocols and procedures could be established – providing a template to build on in follow up studies involving more experimental drugs.
Like I said, I don’t have any of the answers, but this is how I would envisage things moving ahead. And I would love to hear others thoughts on this.
There won’t be any rocks being thrown here – no margin in it. Constructive discussion is what I seek. Plus I’m standing in a glass house.
Many thanks again for the thoughtful comment,
Simon
LikeLike
Simon, such a great blog and a great thing you are doing. Have you heard of HealthUnlocked? They have a Parkinsons Movement section and your blog is frequently cited. I have shared info on your blogs as well. This is another great blog and I love it… Here is a rant/ question:-)
Re trials, I am desperate to get into the best one I can find. I also follow trials such as the NAC trial. I, like many other PWP (People With Parkinson’s), take 2 600 mg capsules a day which mirrors the non infusion component of the study. Is it helping? I don’t know. I began doing this when I learned I had PD, 2 years ago. It vexes me that the reports with conclusions take FOREVER to publish. What about midpoint reports? Where are they? I’d love a midpoint report on israpidine and inosine. Since I am at the 2 year mark (as of 2 weeks ago) since my PD diagnosis, I am a marginal candidate for PASADENA study. I’d love your thoughts for how to get into the best trial and the ability to get data on study updates.
Thanks for listening.
LikeLike
Hi Pamela,
Thanks for your comment and kind words. Yes, I have seen HealthUnlocked (https://healthunlocked.com/). It looks to be a very useful resource. Another good forum for discussing PD research is on Facebook – the Parkinson’s Research Interest Group (https://www.facebook.com/groups/482344755486600).
I appreciate your frustration and anxiety regarding the delay/absence of clinical trials reporting results. It is a real issue for the industry. The issue with providing midpoint reports is that such announcements could adversely affect the outcome of the rest of the study. The participants do not live in a bubble and may hear something in the news about the study they are participating in. That said, the FDA sometimes steps in and halts clinical trials where there is an extremely obvious positive outcome and it is deemed unethical to continue. Those situation usually result in the report being produced rather rapidly.
Regarding the best study to partake in, this is a difficult question to answer as it will depend on your personal medical history and your geographic location. It can now also depend on whether you carry a genetic variant that is associated with Parkinson’s. There are several useful resources available for searching for clinical trials (such as the Michael J Fox Foundation trial finder – https://foxtrialfinder.michaeljfox.org/ – or the Parkinson’s UK Get Involved page – https://www.parkinsons.org.uk/research/get-involved-research).
I hope this helps.
Kind regards,
Simon
LikeLike
Heartfelt thanks, Simon.
LikeLike