|
# # # # In cancer research, scientists have devised methods of extracting samples of blood from patients and then growing certain populations of cells in those samples. The isolated subpopulations of cells can then be manipulated in cell culture, before they are then injected back into the patient. This is a form of immunotherapy – artificially boosting the immune system to target specific disease-related pathology in the body. Recently, researchers have been exploring this alternative form of immunotherapy in the context of Parkinson’s… with some interesting results. In today’s post, we will look at review this new research and consider the implications in terms of future therapies for Parkinson’s. # # # # |
 Source: lls
Source: lls
Some time back, a friend in oncology (cancer) research said to me that “we are about to cure all blood cancers“. It should be noted that this optimistic friend is a “glass is completely full” type.
“How so?” I asked.
“CAR T-cell technology is amazing. Really coming into bloom” they responded.
“What is CAR T-cell technology?” I asked.
They explained that it is a kind of immunotherapy – a method of boosting the immune system to help us fight disease.
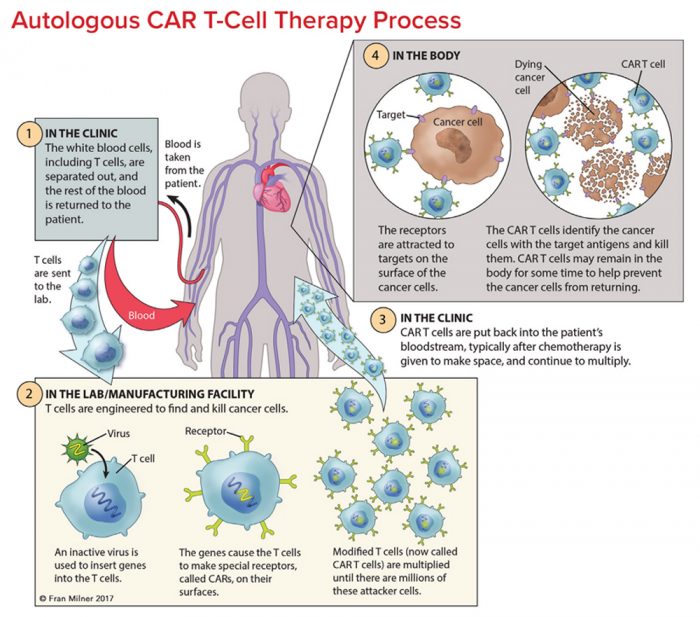
CAR T-cell approaches basically involve removing a sample of blood from a person with cancer, expanding specific populations of those cells in cell culture, genetically manipulating those cells, and then re-introducing them into the body. They also explained that there were lots of different versions of CAR T-cells, with all kinds of potential applications.
“Cool” I said, sounding enthusiastic, but only half understanding what they were saying. My friend is an immunologist, and my summary here is a one sentence version of a 30 minute sermon.
But they are correct.
CAR T-cell technology is achieving really impressive results in cancer (Click here and here to read more about this topic).
Interesting. What does this have to do with Parkinson’s?