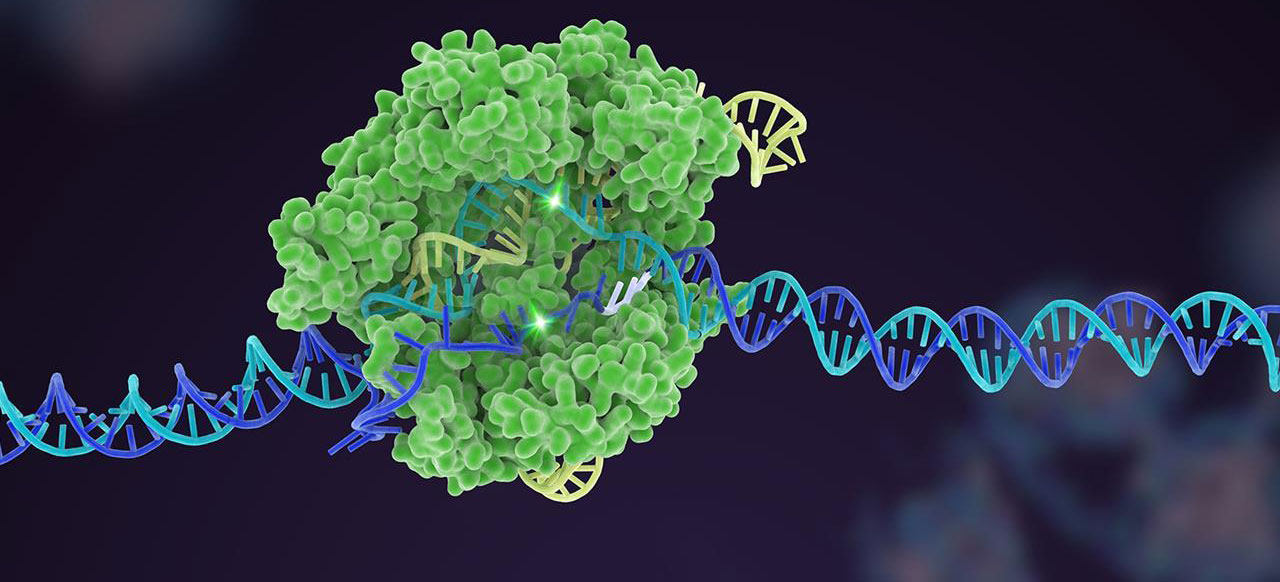
RNA – the usable copy of a section of DNA – has regions called introns that need to be removed before the RNA can be used for the production of protein. The process of removing introns is called splicing.
Recently researchers have noticed that a genetic mutation in a Parkinson’s-associated gene – called DJ-1 – affects the splicing of the associated RNA and this has serious consequences on the activity of the DJ-1 protein.
Interestingly, they were able to pharmacologically rescue the effect, and noticed that DJ-1 might not be the only Parkinson’s-associated gene affected by this splicing error.
In today’s post, we will discuss what splicing is, review the new research, and discuss the wider implications for the Parkinson’s community.
Splice – /splʌɪs/; verb; Meaning: “to combine, interweave”. Origin: 16th century: probably from Middle Dutchsplissen, Similar: braid, plait, entwine, intertwine, interlace, knit Additional/alternative meanings:
1. (From the arts) When two pieces of recorded music – with a similar key and tempo – are combined:
2. (From biology) The process that removes the intervening, non-coding sequences of genes (introns) from pre-mRNA and joins the protein-coding sequences (exons) together in order to enable translation of mRNA into a protein:
Ok, so the first alternative definition about music I understood and the video was helpful, but can you explain the second definition in more detail please?
Recently a Parkinson’s-associated research report was published that was the first of many to come.
It involves the use of a genetic screening experiment that incorporates new technology called ‘CRISPR’.
There is an absolute tidal wave of CRISPR-related Parkinson’s disease research coming down the pipe towards us, and it is important that the Parkinson’s community understands how this powerful technology works.
In today’s post we will look at what the CRISPR technology is, how it works, what the new research report actually reported, and discuss how this technology can be used to tackle a condition like Parkinson’s.
Me and my mother (and yes, the image is to scale). Source: Openclipart
My mother: Simon, what is all this new ‘crispy’ research for Parkinson’s I heard about on the news?
Me: Huh? (I was not really paying attention to the question. Terrible to ignore one’s mother I know, but what can I say – I am the black sheep of the family)
My mother: Yes, something about ‘crispy’ and Parkinson’s.
Me: Oh! You mean CRISPR. Yeah, it’s really cool stuff.
My mother: Ok, well, can you explain it all to me please, this ‘Crisper’ stuff?
Me: Absolutely.
CRISPR.101 (or CRISPR for beginners)
In almost every cell of your body, there is a nucleus.
It is the command centre for the cell – issuing orders and receiving information concerning everything going on inside and around the cell. The nucleus is also a storage bank for the genetic blueprint that provides most of the instructions for making a physical copy of you. Those grand plans are kept bundled up in 23 pairs of chromosomes, which are densely coiled strings of a molecule called Deoxyribonucleic acid (or DNA).
This week a group of scientists have published an article which indicates differences between mice and human beings, calling into question the use of these mice in Parkinson’s disease research.
The results could explain way mice do not get Parkinson’s disease, and they may also partly explain why humans do.
In today’s post we will outline the new research, discuss the results, and look at whether Levodopa treatment may (or may not) be a problem.
Much of our understanding of modern biology is derived from the “lower organisms”.
From yeast to snails (there is a post coming shortly on a snail model of Parkinson’s disease – I kid you not) and from flies to mice, a great deal of what we know about basic biology comes from experimentation on these creatures. So much in fact that many of our current ideas about neurodegenerative diseases result from modelling those conditions in these creatures.
Now say what you like about the ethics and morality of this approach, these organisms have been useful until now. And I say ‘until now’ because an interesting research report was released this week which may call into question much of the knowledge we have from the modelling of Parkinson’s disease is these creatures.
You see, here’s the thing: Flies don’t naturally develop Parkinson’s disease.
Nor do mice. Or snails.
Or yeast for that matter.
So we are forcing a very un-natural state upon the biology of these creatures and then studying the response/effect. Which could be giving us strange results that don’t necessarily apply to human beings. And this may explain our long history of failed clinical trials.
We work with the best tools we have, but it those tools are flawed…
What did the new research report find?
This is the study:
Title: Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease Authors: Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Krüger R, Surmeier DJ, Krainc D Journal: Science, 07 Sept 2017 – Early online publication PMID: 28882997
The researchers who conducted this study began by growing dopamine neurons – a type of cell badly affected by Parkinson’s disease – from induced pluripotent stem (IPS) cells.
On the 27th June, 1997, a research report was published in the prestigious scientific journal ‘Science’ that would change the world of Parkinson’s disease research forever.
And I am not exaggerating here.
The discovery that genetic variations in a gene called alpha synuclein could increase the risk of developing Parkinson’s disease opened up whole new areas of research and eventually led to ongoing clinical trials of potential therapeutic applications.
Todays post recounts the events surrounding the discovery, what has happened since, and we will discuss where things are heading in the future.
In world events, President Bill Clinton was entering his second term, Madeleine Albright became the first female Secretary of State for the USA, Tony Blair became the prime minister of the UK, and Great Britain handed back Hong Kong to China.
In the world of entertainment, author J. K. Rowling’s debut novel “Harry Potter and the Philosopher’s Stone” was published by Bloomsbury, and Teletubbies, South Park, Ally McBeal, and Cold Feet (it’s a British thing) all appeared on TV for the first time, amusing and entertaining the various age groups associated with them.
Musically, rock band Blur released their popular hit song ‘Song 2‘ (released 7th April), “Bitter Sweet Symphony” by the Verve entered the UK charts at number 2 in June, and rapper Notorious B.I.G. was killed in a drive by shooting. Oh, and let’s not forget that “Tubthumping” (also known as “I Get Knocked Down”) by Chumbawamba was driving everybody nuts for its ubiquitous presence.
And at the cinemas, no one seemed to care about anything except a silly movie called Titanic.
The Federal Drug Administration (FDA) in the USA has approved the first drug in 22 years for treating the neurodegenerative condition of Amyotrophic lateral sclerosis (ALS).
The drug is called Edaravone, and it is only the second drug approved for ALS.
In today’s post we’ll discuss what this announcement could mean for Parkinson’s disease.
In 1969, Henry Louis “Lou” Gehrig was voted the greatest first baseman of all time by the Baseball Writers’ Association. He played 17 seasons with the New York Yankees, having signed with his hometown team in 1923.
For 56 years, he held the record for the most consecutive games played (2,130), and he was only prevented from continuing that streak when he voluntarily took himself out of the team lineup on the 2nd May, 1939, after his ability to play became hampered by the disease that now often bears his name. A little more than a month later he retired, and a little less than two years later he passed away.
Amyotrophic lateral sclerosis (or ALS), also known as Lou Gehrig’s disease and motor neuron disease, is a neurodegenerative condition in which the neurons that control voluntary muscle movement die. The condition affects 2 people in every 100,000 each year, and those individuals have an average survival time of two to four years.
In addition to Lou Gehrig, you may have heard of ALS via the ‘Ice bucket challenge‘ (see image in the banner of this post). In August 2014, an online video challenge went viral.
By July 2015, the ice bucket campaign had raised an amazing $115 million for the ALS Association.
Another reason you may have heard of ALS is that theoretical physicist, Prof Stephen Hawking also has the condition:
He was diagnosed with in a very rare early-onset, slow-progressing form of ALS in 1963 (at age 21) that has gradually left him wheel chair bound.
This is very interesting, but what does it have to do with Parkinson’s disease?
Individuals affected by ALS are generally treated with a drug called Riluzole (brand names Rilutek or Teglutik). Approved in December of 1995 by the FDA, this drug increases survival by approximately two to three months.
Until this last week, Riluzole was the only drug approved for the treatment of ALS.
So what happened this week?
On the 5th May, the FDA announced that they had approved a second drug for the treatment of ALS (Click here for the press release).
It is called Edaravone.
What is Edaravone?
Edaravone is a free radical scavenger – a potent antioxidant – that is marketed as a neurovascular protective agent in Japan by Mitsubishi Tanabe Pharma Corporation.
An antioxidant is simply a molecule that prevents the oxidation of other molecules.
Molecules in your body often go through a process called oxidation – losing an electron and becoming unstable. This chemical reaction leads to the production of what we call free radicals, which can then go on to damage cells.
What is a free radical?
A free radical is simply an unstable molecule – unstable because they are missing electrons. They react quickly with other molecules, trying to capture the needed electron to re-gain stability. Free radicals will literally attack the nearest stable molecule, stealing an electron. This leads to the “attacked” molecule becoming a free radical itself, and thus a chain reaction is started. Inside a living cell this can cause terrible damage, ultimately killing the cell.
Antioxidants are thus the good guys in this situation. They are molecules that neutralize free radicals by donating one of their own electrons. The antioxidant don’t become free radicals by donating an electron because by their very nature they are stable with or without that extra electron.
Thus when we say ‘Edaravone is a free radical scavenger’, we mean it’s really good at scavenging all those unstable molecules and stabilising them.
It is an intravenous drug (injected into the body via a vein) and administrated for 14 days followed by 14 days drug holiday.
So, again what has this got to do with Parkinson’s disease?
Well, it is easier to start a clinical trial of a drug if it is already approved for another disease.
And the good news is: Edaravone has been shown to be neuroprotective in several models of Parkinson’s disease.
In this post, we’ll lay out some of the previous research and try to make an argument justifying the clinical testing of Edaravone in Parkinson’s disease
Ok, so what research has been done so far in models of Parkinson’s disease?
The first study to show neuroprotection in a model of Parkinson’s disease was published in 2008:
Title: Role of reactive nitrogen and reactive oxygen species against MPTP neurotoxicity in mice. Authors: Yokoyama H, Takagi S, Watanabe Y, Kato H, Araki T. Journal: J Neural Transm (Vienna). 2008 Jun;115(6):831-42. PMID:18235988
In this first study, the investigators assessed the neuroprotective properties of several drugs in a mouse model of Parkinson’s disease. The drugs included Edaravone (described above), minocycline (antibiotic discussed in a previous post), 7-nitroindazole (neuronal nitric oxide synthase inhibitor), fluvastatin and pitavastatin (both members of the statin drug class).
With regards to Edaravone, the news was not great: the investigators found that Edaravone (up to 30mg/kg) treatment 30 minutes before administering a neurotoxin (MPTP) and then again 90 minutes afterwards had no effect on the survival of the dopamine neurons (compared to a control treatment).
Not a good start for making a case for clinical trials!
This research report, however, was quickly followed by another from an independent group in Japan:
Title: Neuroprotective effects of edaravone-administration on 6-OHDA-treated dopaminergic neurons. Authors: Yuan WJ, Yasuhara T, Shingo T, Muraoka K, Agari T, Kameda M, Uozumi T, Tajiri N, Morimoto T, Jing M, Baba T, Wang F, Leung H, Matsui T, Miyoshi Y, Date I. Journal: BMC Neurosci. 2008 Aug 1;9:75. PMID:18671880 (This article is OPEN ACCESS if you would like to read it)
These researchers did find a neuroprotective effect using Edaravone (both in cell culture and in a rodent model of Parkinson’s disease), but they used a much higher dose than the previous study (up to 250 mg/kg in this study). This increase in dose resulted in a graded increase in neuroprotection – interestingly, these researchers also found that 30mg/kg of Edaravone had limited neuroprotective effects, while 250mg/kg exhibited robust dopamine cell survival and rescued the behavioural/motor features of the model even when given 24 hours after the neurotoxin.
The investigators concluded that “Edaravone might be a hopeful therapeutic option for PD, although several critical issues remain to be solved, including high therapeutic dosage of Edaravone for the safe clinical application in the future”
This results was followed by several additional studies investigating edaravone in models of Parkinson’s disease (Click here, here and here to read more on this). Of particular interest in all of those follow up studies was a report in which Edaravone treatment resulted in neuroprotective in genetic model of Parkinson’s disease:
Title: Edaravone prevents neurotoxicity of mutant L166P DJ-1 in Parkinson’s disease. Authors: Li B, Yu D, Xu Z. Journal: J Mol Neurosci. 2013 Oct;51(2):539-49. PMID:23657982
DJ-1 is a gene that has been associated Parkinson’s disease since 2003. The gene is sometimes referred to as PARK7 (there are now more than 20 Parkinson’s associated genomic regions, which each have a number and are referred to as the PARK genes). Genetic mutations in the DJ-1 gene can result in an autosomal recessive (meaning two copies of the mutated gene are required), early-onset form of Parkinson disease. For a very good review of DJ-1 in the context of Parkinson’s disease, please click here.
The exact function of DJ-1 is not well understood, though it does appear to play a role in helping cells deal with ‘oxidative stress’ – the over-production of those free radicals we were talking about above. Now given that edaravone is a potent antioxidant (reversing the effects of oxidative stress), the researchers conducting this study decided to test Edaravone in cells with genetic mutations in the DJ-1 gene.
Their results indicated that Edaravone was able to significantly reduce oxidative stress in the cells and improve the functioning of the mitochondria – the power stations in each cell, where cells derive their energy. Furthermore, Edaravone was found to reduce the amount of cell death in the DJ-1 mutant cells.
More recently, researchers have begun digging deeper into the mechanisms involved in the neuroprotective effects of Edaravone:
Title: Edaravone leads to proteome changes indicative of neuronal cell protection in response to oxidative stress. Authors: Jami MS, Salehi-Najafabadi Z, Ahmadinejad F, Hoedt E, Chaleshtori MH, Ghatrehsamani M, Neubert TA, Larsen JP, Møller SG. Journal: Neurochem Int. 2015 Nov;90:134-41. PMID:26232623 (This article is OPEN ACCESS if you would like to read it)
The investigators who conducted this report began by performing a comparative two-dimensional gel electrophoresis analyses of cells exposed to oxidative stress with and without treatment of Edaravone.
Um, what is “comparative two-dimensional gel electrophoresis analyses”?
Good question.
Two-dimensional gel electrophoresis analyses allows researchers to determine particular proteins within a given solution. Mixtures of proteins are injected into a slab of gel and they are then separated according to two properties (mass and acidity) across two dimensions (left-right side of the gel and top-bottom of the gel).
A two-dimensional gel electrophoresis result may look something like this:
Two-dimensional gel electrophoresis. Source: Nature
As you can see, individual proteins have been pointed out on the image of this slab of gel.
In comparative two-dimensional gel electrophoresis, two samples of solution are analysed by comparing two slabs of gel that have been injected with protein mix solution from two groups of cells treated exactly the same except for one variable. Each solution gets its own slab of gel, and the differences between the gel product will highlight which proteins are present in one condition versus the other (based on the variable being tested).
In this experiment, the variable was Edaravone.
And when the researchers compared the proteins of Edaravone treated cells with those of cells not treated with Edaravone, they found that the neuroprotective effect of Edaravone was being caused by an increase in a protein called Peroxiredoxin-2.
Now this was a really interesting finding.
You see, Peroxiredoxin proteins are a family (there are 6 members) of antioxidant enzymes. And of particular interest with regards to Parkinson’s disease is the close relationship between DJ-1 (the Parkinson’s associated protein discussed above) and peroxiredoxin proteins (Click here, here, here and here to read more about this).
In addition, there are also 169 research reports dealing with the peroxiredoxin proteins and Parkinson’s disease (Click here to see a list of those reports).
So, what do you think about a clinical trial for Edaravone in Parkinson’s disease?
Are you convinced?
Regardless, it an interesting drug huh?
Are there any downsides to the drug?
One slight issue with the drug is that it is injected via a vein. Alternative systems of delivery, however, are being explored.A biotech company in the Netherlands, called Treeway is developing an oral formulation of edaravone (called TW001) and is currently in clinical development.
Edaravone was first approved for clinical use in Japan on May 23, 2001. With almost 17 years of Edaravone clinical use, a few adverse events including acute renal failure have been noted, thus precautions should be taken with individuals who have a history of renal problems. The most common side effects associated with the drug, however, are: fatigue, nausea, and some mild anxiety.
Click here for a good overview of the clinical history of Edaravone.
So what does it all mean?
The announcement from the FDA this week regarding the approval of Edaravone as a new treatment for ALS represents a small victory for the ALS community, but it may also have a significant impact on other neurodegenerative conditions, such as Parkinson’s disease.
Edaravone is a potent antioxidant agent, which has been shown to have neuroprotective effects in various models of Parkinson’s disease and other neurodegenerative conditions. It could be interesting to now test the drug clinically for Parkinson’s disease. Many of the preclinical research reports indicate that the earlier Edaravone treatment starts, the better the outcomes, so any initial clinical trials should focus on recently diagnosed subjects (perhaps even those with DJ-1 mutations).
The take home message of this post is: given that Edaravone has now been approved for clinical use by the FDA, it may be advantageous for the Parkinson’s community to have a good look at whether this drug could be repurposed for Parkinson’s disease.
It’s just a thought.
The banner for today’s post was sourced from Forbes
A community in New Brunswick (Canada) was recently shocked to discover that a 2 year old boy in their midst had been diagnosed with Parkinson’s disease (Click here to read more).
Yes, you read that correctly, it’s not a typo: a 2 year old boy.
Juvenile-onset Parkinson’s disease is an extremely rare version of the condition we discuss here at the Science of Parkinson’s. It is loosely defined as being ‘diagnosed with Parkinson’s disease under the age of 20’. The prevalence is unknown, but there is a strong genetic component to form of the condition. In today’s post we will review what is known about Juvenile-onset and look at new research about a gene that has recently been discovered to cause a type of Juvenile-onset Parkinson’s disease.
In 1875, Dr Henri Huchard (1844-1910; a French neurologist and cardiologist) described the first case of a child who, at just 3 years of age, presented all the clinical features of Parkinson’s disease. Since that report, there have been many studies detailing the condition that has become known as ‘juvenile-onset Parkinson’s disease’.
What is juvenile-onset Parkinson’s disease?
Basically, it is a form of Parkinson’s disease that affects children and young people under the age of 20. The defining feature is the age of onset. The average age of onset is approximately 12 years of age (with the majority of cases falling between 7 and 16 years) and males are affected by this condition more than females (at a rate of approximately 5:1).
The actual frequency of juvenile-onset parkinson’s is unknown given how rare it is. When researcher look at people with early onset Parkinson’s disease (that is diagnosis before the age of 40; approximately 5% of the Parkinson’s community), they have found that between 0.5 – 5% of that group of people were diagnosed before 20 years of age. This suggests that within just the Parkinson’s community, the frequency of juvenile-onset parkinson’s is at the most 0.25% (or 2.5 people per 1000 people with Parkinson’s). Thus it is obviously a very rare condition.
It is interesting to note that Lewy bodies (the clusters of aggregated protein that classically characterise the brains of people with Parkinson’s disease) are very rare in cases of juvenile-onset parkinson’s disease. To our knowledge there has been only one case of Lewy bodies in juvenile-onset parkinson’s disease (Click here to read more on this). This suggests that the juvenile-onset form of Parkinson’s disease may differ from other forms of the condition in its underlying biology.
Do we know what causes juvenile-onset parkinson’s disease?
There is a very strong genetic component to juvenile-onset parkinson’s disease. In fact, the incidence of Parkinsonism in relatives of people with juvenile-onset parkinson’s disease is higher than in the general public AND in the relatives of people with other forms of Parkinson’s disease.
Genetic mutations in three genes are recognised as causing juvenile-onset Parkinson’s disease. The three genes are known to the Parkinson’s world as they are all PARK genes (genetic variations that are associated with Parkinson’s). Those three genes are:
Parkin (PARK2)
PTEN-induced putative kinase 1 (PINK1 or PARK6)
DJ1 (PARK7)
In juvenile-onset Parkinson’s disease, all of these mutations are associated with autosomal recessive – meaning that two copies of the genetic variation must be present in order for the disease to develop.
Parkin mutations account for the majority of juvenile-onset Parkinson’s disease cases. Affected individuals have a slowly progressing condition that is L-dopa responsive. Dystonia (abnormal muscle tone resulting in muscular spasm and abnormal posture) is very common at the onset of the condition, particularly in the lower limbs.
Can the condition be treated with L-dopa?
The answer is: ‘Yes, but…’
L-dopa (or dopamine replacement) treatment is the standard therapy for alleviating the motor features of Parkinson’s disease.
The majority of people with juvenile-onset parkinson’s respond well to L-dopa, but in the Parkin mutation version individuals will typically begin to experience L-dopa-induced motor fluctuations (dyskinesias) early in that treatment regime.
What research is currently being done on this condition?
Given that cases are so very rare and so few, it is difficult to conduct research on this population of individuals. Most of the research that is being conducted is focused on the genetics underlying the condition.
And recent that research lead to the discovery of a new genetic variation that causes juvenile-onset Parkinson’s disease:
Title: Discovery of a frameshift mutation in podocalyxin-like (PODXL) gene, coding for a neural adhesion molecule, as causal for autosomal-recessive juvenile Parkinsonism. Authors: Sudhaman S, Prasad K, Behari M, Muthane UB, Juyal RC, Thelma BK. Journal: Journal Med Genet. 2016 Jul;53(7):450-6. PMID:26864383 (This article is OPEN ACCESS if you would like to read it)
The researchers who wrote this article were presented with a 10 member Indian family from Aligarh, Uttar Pradesh. Of the 8 children in the family, 3 were affected by Parkinsonian features (tremor, slowness, rigidity and gait problems) that began between 13 and 17 years of age. The researchers conducted DNA sequencing and found that none of the three affected siblings had any of the known Juvenile-onset Parkinson’s disease genetic mutations (specifically, mutations in the genes PARK2, PINK1and DJ1).
They then compared the DNA from the three siblings with the rest of the family and found a genetic variant in a gene called podocalyxin-like (or PODXL). It must be noted that PODXL is a completely novel gene in the world of Parkinson’s disease research, which makes it very interesting. PODXL has never previously been associated with any kind of Parkinson’s disease, though it has been connected with two types of cancer (embryonal carcinoma and periampullary adenocarcinoma).
The researchers then turned to their genetic database of 280 people with Parkinson’s disease have had their genomes sequenced. The researchers wanted to determine if any genetic variants in the PODXL gene were present in other suffers of Parkinson’s disease, but had not been picked up as a major contributing factor. They found three unrelated people with PODXL mutations. All three had classical Parkinson’s features, and were negative for mutations in the Parkin, PINK1 and DJ1 genes.
The researchers concluded that the PODXL gene may be considered as a fourth causal gene for Juvenile-onset Parkinson’s disease, but they indicated that further investigations in other ethnic groups are required.
The banner for today’s post was sourced from ClipArtBest

 Source: Wikibooks
Source: Wikibooks