In 2018, there is one particular clinical trial that I will be watching, because the drug being tested could have a big impact on certain kinds of Parkinson’s.
The clinical trial is focused on people with cancer and they will be treated with a drug called TVB-2640. TVB-2640 is an inhibitor of an enzyme called fatty acid synthase (or FAS).
In today’s post we will discuss why TVB-2640 might be a useful treatment for certain kinds of Parkinson’s.
Mitochondria and their location in the cell. Source: NCBI
Regular readers of this blog are probably getting sick of the picture above.
I use it regularly on this website, because a.) it nicely displays a basic schematic of a mitochondrion (singular), and where mitochondria (plural) reside inside a cell. And b.) a lot of evidence is pointing towards mitochondrial dysfunction in Parkinson’s.
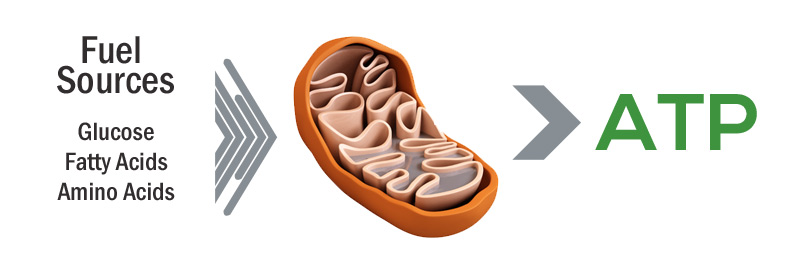
What are mitochondria?
Mitochondria are the power stations of each cell. They help to keep the lights on. Without them, the party is over and the cell dies.
How do they supply the cell with energy?
They convert nutrients from food into Adenosine Triphosphate (or ATP). ATP is the fuel which cells run on. Given their critical role in energy supply, mitochondria are plentiful (some cells have thousands) and highly organised within the cell, being moved around to wherever they are needed.
“Here they make all sorts of cutlery-ware, but especially that of edged-tools, knives, razors, axes, &. and nails; and here the only mill of the sort, which was in use in England for some time was set up, for turning their grindstones, though now ’tis grown more common”
Sheffield has a long history of metal work, thanks largely to its geology: The city is surrounded by fast-flowing rivers and hills containing many of the essential raw materials such as coal and iron ore.
And given this fortunate circumstance and an industrious culture, the city of Sheffield particularly prospered during the industrial revolution of the mid-late 1800s (as is evident from the population growth during that period).
The population of Sheffield over time. Source: Wikipedia
But traditional manufacturing in Sheffield (along with many other areas in the UK) declined during the 20th century and the city has been forced to re-invent itself in the early 21st century. And this time, rather than taking advantage of their physical assets, the city is focusing on its mental resources.
Great. Interesting stuff. Really. But what does this have to do with flies, fish and Parkinson’s disease???
Indeed. Let’s get down to business.
The Sheffield Institute for Translational Neuroscience (SITraN) was officially opened in 2010 by Her Majesty The Queen. It is the first European Institute purpose-built and dedicated to basic and clinical research into Motor Neuron Disease as well as related neurodegenerative disorders such as Parkinson’s and Alzheimer’s disease.
Since its opening, the institute has published some pretty impressive research, particularly in the field of Parkinson’s disease.
We have previously discussed “Pink” flies and their critical role in Parkinson’s research (Click here to read that post).
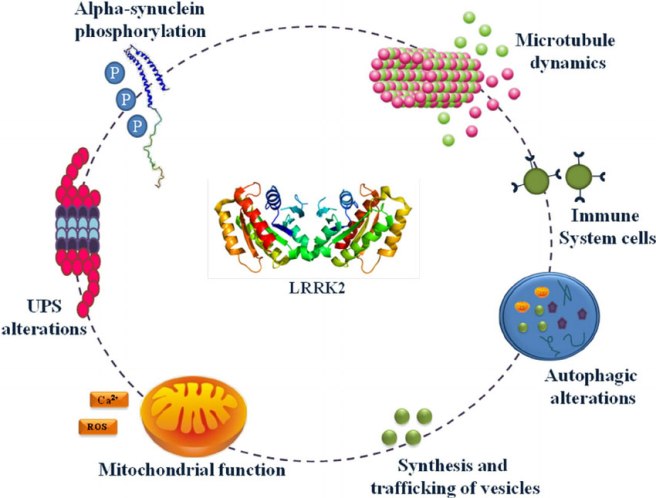
Today we are going to talk about Lrrk2 flies.
What is Lrrk2?
This is Sergey Brin.
He’s a dude.
One of the founders of the search engine company “Google”. Having changed the world, he is now turning his attention to other projects.
One of those other projects is close to our hearts: Parkinson’s disease.
In 1996, Sergey’s mother started experiencing numbness in her hands. Initially it was believed to be RSI (Repetitive strain injury). But then her left leg started to drag. In 1999, following a series of tests, Sergey’s mother was diagnosed with Parkinson’s disease. It was not the first time the family had been affected by the condition: Sergey’s late aunt had also had Parkinson’s disease.
Both Sergey and his mother have had their DNA scanned for mutations that increase the risk of Parkinson’s disease. And they discovered that they were both carrying a mutation on the 12th chromosome, in a gene called PARK8 – one of the Parkinson’s disease associated genes. Autosomal dominant mutations (meaning if you have just one copy of the mutated gene) in the PARK8 gene dramatically increase one’s risk of developing Parkinson’s disease.
PARK8 provides the instructions for making an enzyme called Leucine-rich repeat kinase 2 (or Lrrk2).
Also known as ‘Dardarin‘ (from the Basque word “dardara” which means trembling), Lrrk2 has many functions within a cell – from helping to move things around inside the cell to helping to keep the power on (involved with mitochondrial function).
NOTE: Curiously, mutations in the PARK8 gene are also associated with Crohn’s disease (Click here and here for more on this) – though the mutation is in a different location for PD.
Now, not everyone with this particular mutation will go on to develop Parkinson’s disease, and Sergey has decided that his chances are 50:50. But he does not appear to be taking any chances though. Being one of the founders of a large company like Google, has left Sergey with considerable resources at his disposal. And he has chosen to focus some of those resources on Lrrk2 research (call it an insurance policy). He has done this via considerable donations to groups like the Michael J Fox foundation.
Actor Michael J Fox was diagnosed at age 30. Source: MJFox foundation
So just as Pink flies derive their name from mutations in the Parkinson’s associated Pink1 gene, Lrrk2 flies have mutations in the Lrrk2 gene.
So what have the researchers at Sheffield done with the Lrrk2 flies?
In 2013, the Sheffield researchers published an interesting research report:
Title: Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease Authors: Mortiboys H, Aasly J, Bandmann O. Journal: Brain. 2013 Oct;136(Pt 10):3038-50. PMID:24000005
In this study, the investigators took 2000 drugs (including 1040 licensed drugs and 580 naturally occurring compounds) and conducted a massive screen to identify drugs that could rescue mitochondrial dysfunction in PARK2 (Parkin) mutant cells.
Mitochondria are the power house of each cell. They keep the lights on. Without them, the lights go out and the cell dies.
Mitochondria and their location in the cell. Source: NCBI
In certain genetic forms of Parkinson’s disease (such as those associated with mutations in the PARK2 gene), the mitochondria in cells becomes dysfunctional and may not be disposed of properly (Click here to read our previous post related to this).
In their huge screen of 2000 drugs, the researchers in Sheffield identified 15 drugs that could rescue the mitochondria dysfunction in the PARK2 skins cells. Of those 15 compounds, two were chosen for further functional studies. They were:
Ursocholanic acid
Dehydro(11,12)ursolic acid lactone
Neither ursocholanic acid nor dehydro(11,12)ursolic acid lactone are FDA-licensed drugs. We have little if any information regarding their use in humans. Given this situation, the researchers turned their attention to the chemically related bile acid ‘ursodeoxycholic acid’, which has been in clinical use for more than 30 years.
What is Ursodeoxycholic Acid?
Ursodeoxycholic Acid (or UDCA) is a drug that is used to to improve bile flow and reduce gallstone formation. In the USA it is also known as ‘ursodiol’.
Bile is a fluid that is made and released by your liver, and it stored in the gallbladder. Its function is to help us with digestion. UDCA occurs naturally in bile – it is basically a bile acid and can therefore be useful in dissolving gallstones. UDCA has been licensed for the treatment of patients since 1980. UDCA also reduces cholesterol absorption.
So what did the Sheffield researchers find with UDCA?
The researchers tested UDCA on mitochondrial function in PARK2 skin cells, and they found that the drug rescued the cells. They then tested UDCA on skin cells from people with Parkinson’s disease who had mutations in the PARK8 (Lrrk2) gene (G2019S).
The researchers had previously found impaired mitochondrial function and morphology in skin cells taken from people with PARK8 associated Parkinson’s disease (Click here to read more about this), and other groups had reported similar findings (Click here for more on this).
And when they treated the Lrrk2 cells with UDCA, guess what happened?
UDCA was able to rescue the mitochondrial effect in those cells as well!
Obviously these results excited the Sheffield scientists and they set up a collaboration with researchers at York University and from Norway, to look at the potential of UDCA in rescuing the fate of Lrrk2 flies. The results of that study were published two years ago:
Title: UDCA exerts beneficial effect on mitochondrial dysfunction in Lrrk2 (G2019S) carriers and in vivo. Authors: Mortiboys H, Furmston R, Bronstad G, Aasly J, Elliott C, Bandmann O. Journal: Neurology. 2015 Sep 8;85(10):846-52. PMID:26253449 (This article is OPEN ACCESS if you would like to read it).
The researchers tested UDCA on flies (or drosophila) with specific Lrrk2 mutations (G2019S) display a progressive loss of photoreceptor cell function in their eyes. The mitochondria in the photoreceptor are swollen and disorganised. When the investigators treated the flies with UDCA, they found approximately 70% rescue of the photoreceptor cells function.
The researchers in Sheffield concluded that UDCA has a marked rescue effect on cells from a Parkinson’s disease-associated gene mutation model, and they proposed that “mitochondrial rescue agents may be a promising novel strategy for disease-modifying therapy in Lrrk2-related PD, either given alone or in combination with Lrrk2 kinase inhibitors” (for more information about the Lrrk2 inhibitors they refer, click here).
And the good news regarding this line of research: other research groups have also observed similar beneficial effects with UDCA in models of Parkinson’s disease:
Title: Ursodeoxycholic acid suppresses mitochondria-dependent programmed cell death induced by sodium nitroprusside in SH-SY5Y cells. Authors: Chun HS, Low WC. Journal: Toxicology. 2012 Feb 26;292(2-3):105-12. PMID:22178905
This research group also demonstrated that UDCA could reduce cell death in a cellular model of Parkinson’s disease.
And this study was followed by another one from a different research group, which involved testing UDCA in animals:
Title: Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Authors: Abdelkader NF, Safar MM, Salem HA. Title: Mol Neurobiol. 2016 Mar;53(2):810-7. PMID:25502462
These researchers found UDCA rescued a rodent model of Parkinson’s disease (involving the neurotoxin rotenone). UDCA not only improved mitochondrial performance in the rats, but also demonstrated anti-inflammatory and anti-cell death properties.
Given all this research, the Sheffield researchers are now keen to test UDCA in clinical trials for Parkinson’s disease.
Has anyone tested UDCA in the clinic for Parkinson’s disease?
Not that we are aware of, but two groups are interested in attempting it.
Firstly, the University of Minnesota – Clinical and Translational Science Institute has registered a trial (Click here to read more about this). This trial will not, however, be testing efficacy of the drug on Parkinson’s symptoms. It will focus on measuring UDCA levels in individuals after four weeks of repeated high doses of oral UDCA (50mg/kg/day), and determining the bioenergetic profile and ATPase activity in those participants. Basically, they want to see if UDCA is safe and active in people with Parkinson’s disease.
The CurePD trust (in the UK) is also currently seeking to run a clinical trial for UDCA (Click here for more on this). The group are currently organising the funding for that trial.
EDITOR’S NOTE HERE: Before we move on, the team at the SoPD would like to say that while UDCA is a clinically available drug, it is still experimental for Parkinson’s disease. There is no indication yet that it has beneficial effects in people with Parkinson’s disease. In addition, UDCA is also is known to have side effects, which include flu symptoms, nausea, diarrhea, and back pain. And individuals have been known to have allergic reactions to UDCA treatment (Click here and here for more on the side effects of UDCA). Thus we must impress caution on anyone planning to experiment with this drug. Before attempting any kind of change in a current treatment regime, PLEASE discuss your plans with a medically qualified physician who is familiar with your case history.
Ok, so that was the flies research, what about the fish? And the… uh, tigar?
Yes. The fish are called Zebrafish (or Danio rerio).
They are a tropical freshwater fish that is widely used in biological research.
Biology researchers love these little guys because their genome has been fully sequenced and they has well characterised and testable behaviours. In addition, their development is very rapid (3 months), and its embryos are large and transparent.
And the researchers at Sheffield are using these fish to study Parkinson’s disease.
How did they do that?
Title: TigarB causes mitochondrial dysfunction and neuronal loss in Pink1 deficiency Authors: Flinn LJ, Keatinge M, Bretaud S, Mortiboys H, Matsui H, De Felice E, Woodroof HI, Brown L, McTighe A, Soellner R, Allen CE, Heath PR, Milo M, Muqit MM, Reichert AS, Köster RW, Ingham PW, Bandmann O. Journal: Ann Neurol. 2013 Dec;74(6):837-47.
PMID: 24027110 (This article is OPEN ACCESS if you would like to read it)
Firstly, the group at Sheffield generated zebrafish that had a mutation in the Parkinson’s associated gene ‘PARK6’. This gene provides the plans for the production of a protein called Pink1 (we have previously discussed Pink1 – click here to read more on this).
In normal healthy cells, the Pink1 protein is absorbed by mitochondria and eventually degraded as it is not used. In unhealthy cells, however, this process becomes inhibited and Pink1 starts to accumulate on the outer surface of the mitochondria. Sitting on the surface, it starts grabbing another Parkinson’s associated protein called Parkin. This pairing is a signal to the cell that this particular mitochondria is not healthy and needs to be removed.
Pink1 and Parkin in normal (right) and unhealthy (left) situations. Source: Hindawi
The process by whichmitochondria are removed is called mitophagy. Mitophagy is part of the autophagy process, which is an absolutely essential function in a cell. Without autophagy, old proteins and mitochondria will pile up making the cell sick and eventually it dies. Through the process of autophagy, the cell can break down the old protein, clearing the way for fresh new proteins to do their job.
Think of autophagy as the waste disposal/recycling process of the cell.
Waste material inside a cell is collected in membranes that form sacs (called vesicles). These vesicles then bind to another sac (called a lysosome) which contains enzymes that will breakdown and degrade the waste material. The degraded waste material can then be recycled or disposed of by spitting it out of the cell.
In the case of a PARK6 mutations, Pink1 protein can not function properly with Parkin and the autophagy process breaks down. As a result, the old or unhealthy mitochondria start to pile up in the cell, resulting in the cell getting sick and dying.
Now back to the Zebrafish.
When the Sheffield researchers mutated PARK6 in the zebrafish, they noticed that the fish had a very early and persistent loss of dopamine neurons in their brains. These fish also had enlarged, unhealthy mitochondria and reduced mitochondrial activity.
Given this result, the investigators next wanted to identify which genes have increased or decreased levels of activity as a result of this genetic manipulation. They identified 108 genes that were higher in the PARK6 mutant, and 146 genes had lower activity.
One gene in particular had activity levels 12 times higher in the PARK6 mutant fish than the normal zebrafish.
The name of that gene? TP53-Induced Glycolysis And Apoptosis Regulator (or Tigar).
What is Tigar?
Tigar is a gene that provides the instructions for making a protein that is activated by p53 (also known as TP53).
What does that mean?
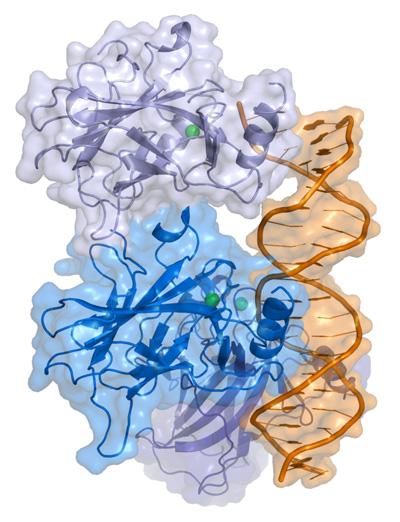
p53 is a protein that has three major functions: controlling cell division, DNA repair, and apoptosis (or cell death). p53 performs these functions as a transcriptional activator (that is a protein that binds to DNA and helps produce RNA (the process of transcription) – see our previous post explaining this).
p53 protein structure, bound to DNA (in gold). Source: Wikipedia
In regulating the cell division, p53 prevents cells from dividing too much and in this role it is known as a tumour suppression – it suppresses the emergence of cancerous tumours. Genetic mutations in the p53 gene result in run away cell division, and (surprise!) as many as 50% of all human tumours contain mutations in the p53 gene.
In DNA repair, p53 is sometimes called “the guardian of the genome” as it prevents mutations and helps to conserve stability in the genome. This function also serves to prevent the development of cancer, by helping to repair potentially cancer causing mutations….and in this role it is known as a tumour suppression. Obviously, if there is a mutation in the p53 gene, less DNA repair will occur – increasing the risk of cancer occurring.
And finally, in cell death, p53 plays a critical role in telling a cell when to die. And (continuing with the cancer theme), if there is a mutation in the p53 gene, fewer cells will be told to die – increasing the risk of cancer occurring. And in this role p53 is known as a tumour suppression.
In normal cells, the levels of p53 protein are usually low. When a cell suffers DNA damage and stress, there is often an increase in the amount of p53 protein. If this increases past a particular threshold, then the cell will be instructed to die.
If you haven’t guessed yet, p53 is a major player inside most cell, and it controls the activity of a lot of genes.
And one of those genes is Tigar.
But what does Tigar actually do?
So we have explained the “TP53-Induced” part of the “TP53-Induced Glycolysis And Apoptosis Regulator” name, let’s now focus on the “Glycolysis And Apoptosis Regulator”
Tigar is an interesting protein because it is an enzyme that primarily functions as a regulator of the breaking down of glucose (“Glycolysis” involves the conversion of glucose into a chemical called pyruvate). In addition to this role, however, Tigar acts in preventing cell death (or apoptosis).
Increased levels of Tigar protects cells from oxidative-stress induced apoptosis, by decreasing the levels of free radicals. In this way, it promotes anti-oxidant activities.
But hang on a second, anti-oxidant activity should be good for the cell right? Why are the dopamine cells are dying if Tigar levels are increasing in the PARK6 mutants?
Fantastic question!
The answer: TIGAR is also a negative regulator of a process called mitophagy. As we discussed above, mitophagy is the process of removing mitochondria by autophagy. Increases in the levels of TIGAR blocks mitophagy in a cell, and results in an increased number of swollen and unhealthy mitochondria in those cells (Click here to read more about this). These swollen mitochondria are comparable to the enlarged mitochondria identified the PARK6 zebrafish by the Sheffield researchers.
And the researchers believe that this may be the cause of the cell death in the PARK6 zebrafish – the double impact of PARK6 and Tigar induced problems with mitophagy.
NOTE: Problems with mitophagy is believed to be an important mechanism in the development of early-onset Parkinson’s disease (Click here for a recent review on this)
Ok, and what did the Sheffield researchers do next?
Given that there was such a huge increase in Tigar levels in the PARK6 zebrafish, the investigators decided to reduce Tigar levels in the PARK6 zebrafish to see what impact this would have on the fish (and their mitochondria).
Remarkably, reductions of Tigar levels resulted in complete rescue of the dopamine neurons in the PARK6 fish. It also increased mitochondrial activity in those cells, and reduced the activation of the microglia cells, which can also play a role in the removal of sick cells in the brain.
The researchers concluded that the results demonstrate that TIGAR is “a promising novel target for disease‐modifying therapy in Pink1‐related Parkinson’s disease”.
And what are the researchers planning to do next with Tigar?
Prof Oliver Bandmann, the senior scientist who ran the study, has said that they “need to finish studying TIGAR levels in the brains of people with Parkinson’s and want to better understand how this protein is involved in maintaining the cell batteries – called ‘mitochondria'” (Source).
Our guess is that the group will also be conducting studies looking at Tigar reduction in rodent models of Parkinson’s disease to determine if this is a viable target in mammals. If Tigar reduction in rodents is found to be effective, the researchers will probably turn their attention to drug screening studies to identify currently available drugs that can reduce the activity of Tigar. Such a drug would provide us with yet another potential treatment for Parkinson’s disease.
We’ll be keeping an eye out for these pieces of research.
This is all very interesting. What does the future hold for Parkinson’s research in Sheffield?
The goal of the company – the first of its kind – is to combine world-leading research from the University with funding and expertise from the charity to help develop revolutionary drugs for Parkinson’s disease.
What is virtual about it? The biotech won’t be building its own labs, employing a team of specialist laboratory scientists, or buying any high-tech equipment (which would all be incredibly expensive). Rather they will form partnerships with groups that do specific tasks the best.
Here is a video of Dr Author Roach (director of Research at Parkinson’s UK) explaining the idea behind this endeavour:
By seeking a collaboration with Sheffield in the creation of a spin-out biotech company, Parkinson’s UK is not only acknowledging Sheffield’s track record, but also making an investment in their future research. While we cannot be entirely sure of what the long-term future holds for Parkinson’s research in Sheffield, we do know that Keapstone will be an important aspect of it in the immediate future.
Could this be a model for the future of Parkinson’s disease research? Only time will tell. We will have a closer look at Keapstone Therapeutics in an upcoming post.
Click here to learn more about the virtual biotech project.
So what does it all mean?
In 2017, we here at the SoPD have decided to begin highlighting some of the Parkinson’s disease research centres as an addition feature on the blog. We have not been approached by the research group in Sheffield or the University itself, and our selection of this city as our first case study was based purely on the fact that we really like what is happening there with regards to Parkinson’s research!
The research group in Sheffield has undertaken multiple lines of research which could potentially providing us with several novel treatment options for Parkinson’s disease. These lines of research have focused not only on clinically available drugs, but also identifying novel targets. We like what they are doing and will keep a close eye on progress there.
And over the next year we will select additional centres of Parkinson’s research based on the same criteria (us liking what they are doing). Our next case study will be the Van Andel Research Institute in Grand Rapids, Michigan (we would hate to be accused of having a UK bias).
EDITORIAL NOTE: Under absolutely no circumstances should anyone reading the material on this website consider it medical advice. The information provided here is for educational purposes only. Before considering or attempting any change in your treatment regime, PLEASE consult with your doctor or neurologist. While some of the drugs discussed on this website are clinically available, they may have serious side effects. We urge caution and professional consultation before altering any treatment regime. SoPD can not be held responsible for any actions taken based on the information provided here.
A community in New Brunswick (Canada) was recently shocked to discover that a 2 year old boy in their midst had been diagnosed with Parkinson’s disease (Click here to read more).
Yes, you read that correctly, it’s not a typo: a 2 year old boy.
Juvenile-onset Parkinson’s disease is an extremely rare version of the condition we discuss here at the Science of Parkinson’s. It is loosely defined as being ‘diagnosed with Parkinson’s disease under the age of 20’. The prevalence is unknown, but there is a strong genetic component to form of the condition. In today’s post we will review what is known about Juvenile-onset and look at new research about a gene that has recently been discovered to cause a type of Juvenile-onset Parkinson’s disease.
In 1875, Dr Henri Huchard (1844-1910; a French neurologist and cardiologist) described the first case of a child who, at just 3 years of age, presented all the clinical features of Parkinson’s disease. Since that report, there have been many studies detailing the condition that has become known as ‘juvenile-onset Parkinson’s disease’.
What is juvenile-onset Parkinson’s disease?
Basically, it is a form of Parkinson’s disease that affects children and young people under the age of 20. The defining feature is the age of onset. The average age of onset is approximately 12 years of age (with the majority of cases falling between 7 and 16 years) and males are affected by this condition more than females (at a rate of approximately 5:1).
The actual frequency of juvenile-onset parkinson’s is unknown given how rare it is. When researcher look at people with early onset Parkinson’s disease (that is diagnosis before the age of 40; approximately 5% of the Parkinson’s community), they have found that between 0.5 – 5% of that group of people were diagnosed before 20 years of age. This suggests that within just the Parkinson’s community, the frequency of juvenile-onset parkinson’s is at the most 0.25% (or 2.5 people per 1000 people with Parkinson’s). Thus it is obviously a very rare condition.
It is interesting to note that Lewy bodies (the clusters of aggregated protein that classically characterise the brains of people with Parkinson’s disease) are very rare in cases of juvenile-onset parkinson’s disease. To our knowledge there has been only one case of Lewy bodies in juvenile-onset parkinson’s disease (Click here to read more on this). This suggests that the juvenile-onset form of Parkinson’s disease may differ from other forms of the condition in its underlying biology.
Do we know what causes juvenile-onset parkinson’s disease?
There is a very strong genetic component to juvenile-onset parkinson’s disease. In fact, the incidence of Parkinsonism in relatives of people with juvenile-onset parkinson’s disease is higher than in the general public AND in the relatives of people with other forms of Parkinson’s disease.
Genetic mutations in three genes are recognised as causing juvenile-onset Parkinson’s disease. The three genes are known to the Parkinson’s world as they are all PARK genes (genetic variations that are associated with Parkinson’s). Those three genes are:
Parkin (PARK2)
PTEN-induced putative kinase 1 (PINK1 or PARK6)
DJ1 (PARK7)
In juvenile-onset Parkinson’s disease, all of these mutations are associated with autosomal recessive – meaning that two copies of the genetic variation must be present in order for the disease to develop.
Parkin mutations account for the majority of juvenile-onset Parkinson’s disease cases. Affected individuals have a slowly progressing condition that is L-dopa responsive. Dystonia (abnormal muscle tone resulting in muscular spasm and abnormal posture) is very common at the onset of the condition, particularly in the lower limbs.
Can the condition be treated with L-dopa?
The answer is: ‘Yes, but…’
L-dopa (or dopamine replacement) treatment is the standard therapy for alleviating the motor features of Parkinson’s disease.
The majority of people with juvenile-onset parkinson’s respond well to L-dopa, but in the Parkin mutation version individuals will typically begin to experience L-dopa-induced motor fluctuations (dyskinesias) early in that treatment regime.
What research is currently being done on this condition?
Given that cases are so very rare and so few, it is difficult to conduct research on this population of individuals. Most of the research that is being conducted is focused on the genetics underlying the condition.
And recent that research lead to the discovery of a new genetic variation that causes juvenile-onset Parkinson’s disease:
Title: Discovery of a frameshift mutation in podocalyxin-like (PODXL) gene, coding for a neural adhesion molecule, as causal for autosomal-recessive juvenile Parkinsonism. Authors: Sudhaman S, Prasad K, Behari M, Muthane UB, Juyal RC, Thelma BK. Journal: Journal Med Genet. 2016 Jul;53(7):450-6. PMID:26864383 (This article is OPEN ACCESS if you would like to read it)
The researchers who wrote this article were presented with a 10 member Indian family from Aligarh, Uttar Pradesh. Of the 8 children in the family, 3 were affected by Parkinsonian features (tremor, slowness, rigidity and gait problems) that began between 13 and 17 years of age. The researchers conducted DNA sequencing and found that none of the three affected siblings had any of the known Juvenile-onset Parkinson’s disease genetic mutations (specifically, mutations in the genes PARK2, PINK1and DJ1).
They then compared the DNA from the three siblings with the rest of the family and found a genetic variant in a gene called podocalyxin-like (or PODXL). It must be noted that PODXL is a completely novel gene in the world of Parkinson’s disease research, which makes it very interesting. PODXL has never previously been associated with any kind of Parkinson’s disease, though it has been connected with two types of cancer (embryonal carcinoma and periampullary adenocarcinoma).
The researchers then turned to their genetic database of 280 people with Parkinson’s disease have had their genomes sequenced. The researchers wanted to determine if any genetic variants in the PODXL gene were present in other suffers of Parkinson’s disease, but had not been picked up as a major contributing factor. They found three unrelated people with PODXL mutations. All three had classical Parkinson’s features, and were negative for mutations in the Parkin, PINK1 and DJ1 genes.
The researchers concluded that the PODXL gene may be considered as a fourth causal gene for Juvenile-onset Parkinson’s disease, but they indicated that further investigations in other ethnic groups are required.
The banner for today’s post was sourced from ClipArtBest















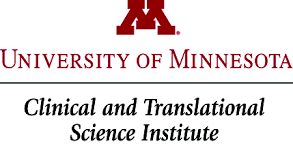











{kind=link}