
|
# # # # Amyotrophic lateral sclerosis (or ALS) is the third most common neurodegenerative condition. It is characterised by the loss of motor neurons, which leads to loss of muscle control. As with Parkinson’s, there is no cure for ALS, and there are only two FDA approved therapies for the condition. Recently, a biotech company – called Amylyx Pharmaceuticals – announced positive Phase II clinical trial results with their experimental combination therapy AMX0035. In today’s post, we will discuss what ALS is, explore the results of the AMX0035 trial, and consider why this could be an important development for Parkinson’s as well. # # # # |

Lou Gehrig. Source: NBC
In 1969, Henry Louis “Lou” Gehrig was voted the greatest first baseman of all time by the Baseball Writers’ Association. During his career, he played 17 seasons with the New York Yankees, having signed with his hometown team in 1923.
For 56 years, he held the record for the most consecutive games played (2,130), and he was only prevented from continuing that streak when he voluntarily took himself out of the team lineup on the 2nd May, 1939, after his ability to play became hampered.
A little more than a month later (at age 36) he retired from the game – his farewell being capped off by his iconic “Luckiest man on the face of the Earth” speech:
And sadly, less than two years later he passed away from the disease that now bears his name: Lou Gehrig’s disease.
Or as it is more commonly known as motor neuron disease.
What exactly is motor neuron disease?
Motor neuron disease – or more correctly amyotrophic lateral sclerosis (ALS) – is a neurodegenerative condition in which the neurons that control voluntary muscle movement die. The condition affects 2 people in every 100,000 each year, and once diagnosed those individuals have an average survival time of two to four years.
 ALS in a nutshell. Source: Walkforals
ALS in a nutshell. Source: Walkforals
In addition to Lou Gehrig, you may have heard of ALS via the ‘Ice bucket challenge‘. In August 2014, an online video challenge went viral:
By July 2015, the ice bucket campaign had raised an amazing $115 million for the ALS Association. And some of these raised funds were used to support the clinical trial we will be discussing in today’s post.
Another reason you may have heard of ALS is that theoretical physicist, Prof Stephen Hawking also has the condition:

Prof Stephen Hawking. Source: BBC
He was diagnosed with in a very rare early-onset, slow-progressing form of ALS in 1963 (at age 21) that gradually left him wheel chair bound.
What causes ALS?
The cause of ALS is unknown, but the condition is characterised by the gradual degeneration and loss of motor neurons.
What are motor neurons?
Motor neurons are neurons that are involved with instructing the muscles of our body. They are typically divided into two types based on their location in the central nervous system.
There are the upper motor neurons which reside in the motor cortex of the brain and they send their branches (or axons) down through the spinal cord. And these axons end on lower motor neurons (the second type) which are located throughout the spinal cord. Lower motor neurons reach out from the spinal cord and make contact with the muscles of our bodies.
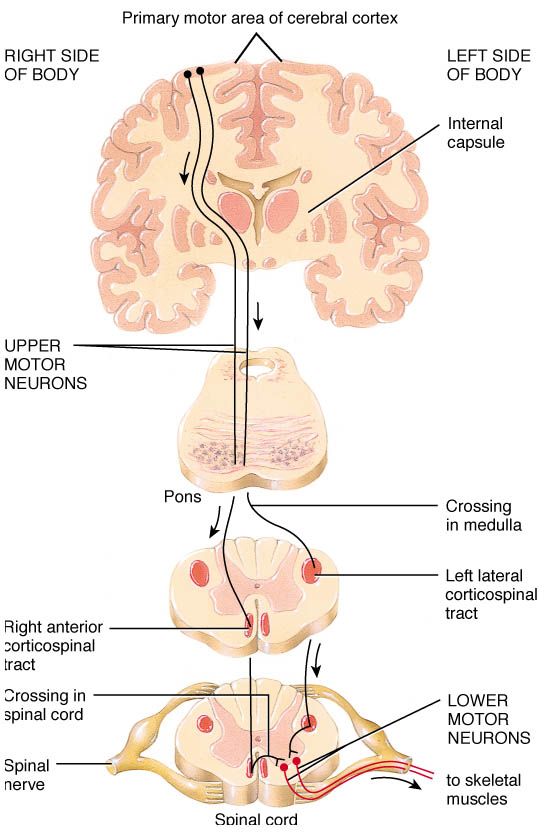 Motor neurons. Source: Pinterest
Motor neurons. Source: Pinterest
If nothing I have written above makes any sense, this video provides a nice basic introduction to what motor neurons are:
And this next video gives a more indepth explanation of motor neurons for those looking for more information:
The loss of motor neurons with ALS causes a loss of muscle control. There are three stages to ALS (Source):
- A gradual development of muscle weakness, tightness, cramping, or twitching. This stage is also associated with muscle loss (or atrophy).
- Muscle weakness and atrophy progresses towards paralysis.
- Most voluntary muscle activity is lost and the sufferer becomes paralyzed. With this stage eating, speaking, and breathing become compromised.
Dementia is not a common feature of the condition, and as a result most individuals affected by ALS retain their higher cognitive abilities and are fully aware of their progressive loss of muscular function.
|
# RECAP #1: Motor neuron disease (also known as ALS) is a neurodegenerative condition, characterised by the gradual loss of motor neurons which leave the sufferer progressively becoming more paralysed. Motor neurons are the cells in the central nervous system that make muscle activity occur. # |
Are there any treatments for ALS?
To date, the U.S. Food and Drug Administration (FDA) has approved four treatments for slowing the progression of ALS:
- Rilutek (riluzole tablet)
- Tiglutik (riluzole suspension)
- Exservan (riluzole oral film)
- Radicava (edaravone)
And yes, as you may have notice, three of these therapies are based on the same molecule (riluzole). Radicava (a potent antioxidant) was only recently approved in 2017 (Click here for the press release). So in theory, there are only 2 approved treatments for ALS.
As a result of these very limited options, the ALS community have been desperate for a new treatment that might shift the needle in terms of slowing progression.
Recently the ALS community received some positive news with the publication of this report:
 Title: Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis.
Title: Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis.
Authors: Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, Karam C, Caress JB, Owegi MA, Quick A, Wymer J, Goutman SA, Heitzman D, Heiman-Patterson T, Jackson CE, Quinn C, Rothstein JD, Kasarskis EJ, Katz J, Jenkins L, Ladha S, Miller TM, Scelsa SN, Vu TH, Fournier CN, Glass JD, Johnson KM, Swenson A, Goyal NA, Pattee GL, Andres PL, Babu S, Chase M, Dagostino D, Dickson SP, Ellison N, Hall M, Hendrix K, Kittle G, McGovern M, Ostrow J, Pothier L, Randall R, Shefner JM, Sherman AV, Tustison E, Vigneswaran P, Walker J, Yu H, Chan J, Wittes J, Cohen J, Klee J, Leslie K, Tanzi RE, Gilbert W, Yeramian PD, Schoenfeld D, Cudkowicz ME.
Journal: N Engl J Med. 2020 Sep 3;383(10):919-930.
PMID: 32877582
In this study, the researchers conducted a Phase II clinical trial that involved 137 people diagnosed with recently diagnosed ALS (<18 months) being randomly assigned to receive either AMX0035 (89 participants) or placebo (48 participants – Click here to read more about the details of the study).
What is AMX0035?
AMX0035 is a combination therapy being developed by the biotech company Amylyx Pharmaceuticals.

AMX0035 involves the combination of sodium phenylbutyrate and taurursodiol.
What does that mean? What are sodium phenylbutyrate and taurursodiol?
Sodium phenylbutyrate is an FDA approved medication that is used to treat urea cycle disorders. The urea cycle converts highly toxic ammonia in the body to urea which can then be excreted. Approximately one in 35,000 people will be diagnosed with a urea cycle disorder, which results in a build up of nitrogen waste in the blood leading to high levels of ammonia. This accumulation can lead to mental retardation and early death. Sodium phenylbutyrate allows the kidneys to excrete excess nitrogen in place of urea.
In cells, sodium phenylbutyrate has been reported to display neuroprotective properties (via inhibit endoplasmic reticulum stress and histone deacetylation).
This has been demonstrated in models of ALS:
 Title: Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice.
Title: Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice.
Authors: Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH Jr, Ferrante RJ.
Journal: J Neurochem. 2005 Jun;93(5):1087-98.
PMID: 15934930 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers reported that treating a genetically engineered mouse model of ALS (G93A mice) with sodium phenylbutyrate (PBA in the graphs below; PBS is a saline placebo solution) significantly extended survival, improved behavioural features, and rescued neuropathological damage.
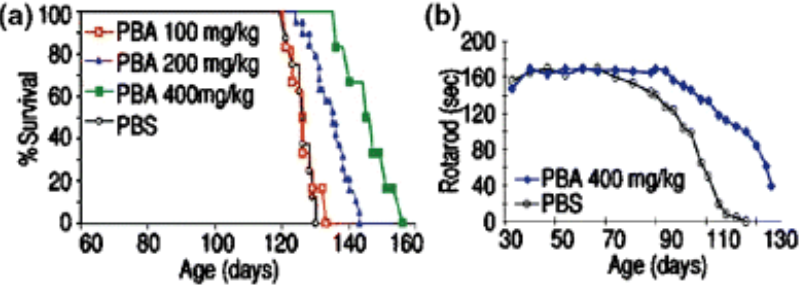 Source: Wiley
Source: Wiley
Similar beneficial effects have been reported in other preclinical studies (Click here and here to read about other examples).
Has sodium phenylbutyrate been clinically tested in ALS before?
Yes it has.
The results of a small Phase II study of sodium phenylbutyrate in individuals with ALS was published in 2009:
 Title: Phase 2 study of sodium phenylbutyrate in ALS.
Title: Phase 2 study of sodium phenylbutyrate in ALS.
Authors: Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH Jr, Zhang H, Schoenfeld DA, Shefner J, Matson S, Matson WR, Ferrante RJ; Northeast ALS and National VA ALS Research Consortiums.
Journal: Amyotroph Lateral Scler. 2009 Apr;10(2):99-106.
PMID: 18688762
In this study, the researchers treated 40 participants at eight research sites in an open-label study of sodium phenylbutyrate in ALS. They found that the doses used (from 9 up to 21 g/day) was safe and well tolerable during the 20-week treatment period, and there were indications of treatment effects (Histone acetylation decreased by approximately 50% during the study).
This result provided support and justification for a larger and longer assessment of sodium phenylbutyrate in ALS.
Ok, what about taurursodiol? What is that?
Taurursodiol is another name for Tauroursodeoxycholic acid (or TUDCA). It is the taurine conjugate form of UDCA – this means that TUDCA is created when a molecule of the organic compound taurine is added to the UDCA structure (see image below).
 The difference between UDCA (top) and TUDCA (bottom). Source: Anabolicminds
The difference between UDCA (top) and TUDCA (bottom). Source: Anabolicminds
TUDCA is approved by the US FDA for treatment of primary biliary cholangitis – a long-term liver condition in which the bile ducts in the liver become damaged.
We have previously discussed both UDCA and TUDCA several times at length on the SoPD (Click here to read a previous SoPD post about both). And UDCA is currently being clinically tested in individuals with Parkinson’s in the “UP-study” – a clinical trial of 30 individuals here in the UK (Click here to read more about this) – and has been tested in a PD cohort in Minnesota (Click here to read a SoPD post about that study).
Has TUDCA ever been clinically tested in ALS before?
Yes it has.
This is the published results of that study:
 Title: Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis.
Title: Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis.
Authors: Elia AE, Lalli S, Monsurrò MR, Sagnelli A, Taiello AC, Reggiori B, La Bella V, Tedeschi G, Albanese A.
Journal: Eur J Neurol. 2016 Jan;23(1):45-52.
PMID: 25664595 (This article is OPEN ACCESS if you would like to read it)
In this study, the researchers conducted a double‐blind, placebo-controlled clinical study on 34 people with ALS (being treated with riluzole) who were randomly assigned take either TUDCA or a placebo twice daily for 54 weeks.
The investigators found that TUDCA was well tolerated – there were no differences between the two groups for adverse events. And the proportion of responders (those who showed improvements) was higher in the TUDCA-treated group (87%) than in the placebo-treated group (this result is very similar to a previous 3 month clinical trial of UDCA in ALS – click here to read more about this). While the study suggests a positive trend regarding efficacy, it was too small to indicate whether TUDCA is demonstrating a statistically significant beneficial effect.
As with the sodium phenylbutyrate trial, the researchers who conducted this study concluded that the results justified a longer, larger trial of TUDCA in ALS.
|
# # RECAP #2: Sodium phenylbutyrate and TUDCA are clinically available therapies that have been shown to have beneficial properties in models of ALS, and to be well tolerated in clinical trials in ALS. Both agents have also been clinically tested in people diagnosed with ALS and found to be safe and well tolerated. # # |
Ok, so what were the results of the new clinical trial of AMX0035 (combination of sodium phenylbutyrate and TUDCA)?
The clinical trial was called the “CENTAUR” study, and the results have been published in two reports.
The first report was published in prestigious The New England Journal of Medicine:
Title: Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis.
Authors: Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, Karam C, Caress JB, Owegi MA, Quick A, Wymer J, Goutman SA, Heitzman D, Heiman-Patterson T, Jackson CE, Quinn C, Rothstein JD, Kasarskis EJ, Katz J, Jenkins L, Ladha S, Miller TM, Scelsa SN, Vu TH, Fournier CN, Glass JD, Johnson KM, Swenson A, Goyal NA, Pattee GL, Andres PL, Babu S, Chase M, Dagostino D, Dickson SP, Ellison N, Hall M, Hendrix K, Kittle G, McGovern M, Ostrow J, Pothier L, Randall R, Shefner JM, Sherman AV, Tustison E, Vigneswaran P, Walker J, Yu H, Chan J, Wittes J, Cohen J, Klee J, Leslie K, Tanzi RE, Gilbert W, Yeramian PD, Schoenfeld D, Cudkowicz ME.
Journal: N Engl J Med. 2020 Sep 3;383(10):919-930.
PMID: 32877582
In this study, the researchers conducted a Phase II clinical trial that involved 137 people diagnosed with recently diagnosed ALS (<18 months). They were randomly assigned in a 2:1 ratio to receive either AMX0035 (89 participants) or placebo (48 participants) for 24 weeks. The primary endpoint of the study (the predetermined measure of success) was the rate of decline in the total score on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (which has a range of 0 to 48 – with higher scores indicating better function).
At the end of the treatment period, the researchers found that the median rate of change in the ALSFRS-R score was -1.24 points per month in the AMX0035-treated group compared with -1.66 points per month in the placebo-treated cohort, which indicated a statistically significant reduction in progression (P=0.03).
In the second report, the researchers examined overall survival in participants who took part in the study:
 Title: Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis.
Title: Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis.
Authors: Paganoni S, Hendrix S, Dickson SP, Knowlton N, Macklin EA, Berry JD, Elliott MA, Maiser S, Karam C, Caress JB, Owegi MA, Quick A, Wymer J, Goutman SA, Heitzman D, Heiman-Patterson TD, Jackson CE, Quinn C, Rothstein JD, Kasarskis EJ, Katz J, Jenkins L, Ladha S, Miller TM, Scelsa SN, Vu TH, Fournier CN, Glass JD, Johnson KM, Swenson A, Goyal NA, Pattee GL, Andres PL, Babu S, Chase M, Dagostino D, Hall M, Kittle G, Eydinov M, McGovern M, Ostrow J, Pothier L, Randall R, Shefner JM, Sherman AV, St Pierre ME, Tustison E, Vigneswaran P, Walker J, Yu H, Chan J, Wittes J, Yu ZF, Cohen J, Klee J, Leslie K, Tanzi RE, Gilbert W, Yeramian PD, Schoenfeld D, Cudkowicz ME.
Journal: Muscle Nerve. 2020 Oct 16. Online ahead of print.
PMID: 33063909
Between March 2018 and September 2019, 90 individuals who took part in the CENTAUR study went on to an open-label extension trial (The Centaur Open Label Extension Study – click here to read more about this).
From the results of that study, the investigators found that the median overall survival period in the AMX0035-treated group was 25 months, compared with just 18.5 months among the placebo-treated cohort, which was also a statistically significant result (P=0.023).
By starting AMX0035-treatment at the start of the trial, participants lived 6.5-months longer compared with the placebo-treated group.
It should be noted that on the secondary measures used in the study there was no statistically significant differences between the two groups, but regardless it is an impressive set of results – that were partly supported by funding from ice bucket challenge fund raising effort (mentioned above). Given the urgent need for new therapies in ALS, it will be interesting to see what the regulators require next for AMX0035 to be made available to the ALS community.
|
# # # RECAP #3: The biotech company Amylyx Pharmaceuticals has developed a combination of sodium phenylbutyrate and TUDCA, called AMX0035. In a Phase II clinical trial in ALS, AMX0035 was found to significantly enhance the survival rate of those treated with this experimental therapy. # # # |
Have sodium phenylbutyrate and TUDCA been tested in models of Parkinson’s?
Yes, they have.
Sodium phenylbutyrate has been reported to increase levels of the Parkinson’s-associated protein DJ-1:
 Title: Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease.
Title: Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease.
Authors: Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR.
Journal: J Biol Chem. 2011 Apr 29;286(17):14941-51.
PMID: 21372141 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers conducted a screening study, evaluating a number of small molecules to identify one that could elevate levels of DJ-1. Their studies found that phenylbutyrate increased levels of DJ-1 in the brain by 260%. They also reported that phenylbutyrate treatment resulted in neuroprotection in both a neurotoxin-based mouse model of PD (MPTP) and a transgenic mouse model of PD (Y39C alpha synuclein). Interestingly, phenylbutyrate treatment reduced levels of alpha synuclein protein aggregation and improved cognitive deficits in aged transgenic mice.
Similar preclinical results have been reported by other independent research groups (Click here, here, here, and here to read examples of this).
NOTE: Eagle eyed readers will notice that sodium phenylbutyrate was mentioned in the previous SoPD post discussing splicing issues associated with genetic variants in the DJ-1 gene – it was referred to as phenylbutyric acid and was used in combination with RECTAS (Click here to read that post).
And as I mentioned above, we have previously discussed both TUDCA several times on the SoPD (Click here to read a previous SoPD post about it). Most recently, we looked at this report: Title: Tauroursodeoxycholic Acid Improves Motor Symptoms in a Mouse Model of Parkinson’s Disease
Title: Tauroursodeoxycholic Acid Improves Motor Symptoms in a Mouse Model of Parkinson’s Disease
Authors: Rosa AI, Duarte-Silva S, Silva-Fernandes A, Nunes MJ, Carvalho AN, Rodrigues E, Gama MJ, Rodrigues CMP, Maciel P, Castro-Caldas M.
Journal: Mol Neurobiol 2018 Apr 12.
PMID: 29651747 (this report is OPEN ACCESS if you would like to read it)
In this study, the researchers conducted an extremely extensive analysis of behaviour to better characterise the mouse model of Parkinson’s (MPTP) that they were using. And after this thorough investigation, they looked to see which aspect of movement they were rescuing with TUDCA treatment. They found that TUDCA administration – either before or after administering the neurotoxin MPTP – significantly improved gait quality, swimming behaviour, and decreased foot dragging.
Importantly, when the investigators looked at the brains of these mice, they found that treating the mice with TUDCA either before or after the neurotoxin reduced levels of a protein called PARIS (see graph below).
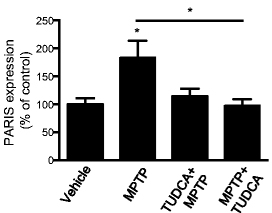 Source: Springer
Source: Springer
We have recently discussed this protein PARIS in the context of Parkinson’s here on the SoPD (Click here to read that post). Increased levels of PARIS are observed in dying dopamine, and reducing levels of PARIS has been found to be neuroprotective. There have been strenuous efforts to find inhibitors of PARIS (and these researchers may have just found one!).
The investigators concluded that the results of this study “should contribute to a subsequent clinical trial in humans and future validation of the therapeutic application of this bile acid in Parkinson’s”.
Have sodium phenylbutyrate and TUDCA been clinically tested in Parkinson’s?
The answer here is: sort of.
A Phase I clinical trial of phenylbutyrate in individuals with Parkinson’s has been conducted at the University of Colorado by the researchers who conducted the DJ-1 study mentioned above (Click here to read more about the trial). It started back in 2014, and we are still waiting to hear any news about the results.
And while TUDCA has not been clinically tested in Parkinson’s (happy to be corrected on this if I am wrong), closely related UDCA has been (TUDCA is broken down into UDCA once it is in the body, giving it better bioavailability compared to UDCA). As I mentioned above, UDCA is currently being clinically tested in individuals with Parkinson’s in the “UP-study” – a clinical trial of 30 individuals here in the UK (Click here to read more about this) and has been tested in a PD cohort in Minnesota (Click here to read a SoPD post about that study).
Is Amylyx Pharmaceuticals planning to test AMX0035 in Parkinson’s?
This is a really good question.
And I am sure that the company would be interested in exploring this idea, but no plans to do so have been announced yet (happy to be corrected on this).
Has AMX0035 been clinically tested in any other neurodegenerative conditions?
Yes.
In August 2018, Amylyx Pharmaceuticals began a Phase 2 clinical trial in Alzheimer’s – the study is called “PEGASUS”. The study is enrolling 100 participants who have had a biomarker-confirmed diagnosis of probable Alzheimer’s disease or mild cognitive impairment. Those participants have been randomised (on a 3:2 ratio) to daily treatment of either AMX0035 or placebo for 24 weeks.
The study is being conducted across 10 sites in the U.S., and the primary endpoint of the study (the main measure) is safety (Click here to read more about the details of the study). On the 9th November (2020) Amylyx announcesd that the last participant in the study has completed their final study visit in PEGASUS trial, and that the topline results of the trial are expected to be announced in the first half of 2021.
So what does it all mean?
There is an interesting backstory to Amylyx Pharmaceuticals (Click here to read the STATnews version). It was started by co-founders Josh Cohen and Justin Klee while they were sitting in a dorm room at Brown University in 2013.
 Justin Klee and Josh Cohen. Source: STAT
Justin Klee and Josh Cohen. Source: STAT
They were obsessing over how to stop neurons from dying and after much literature searching, decided on a combination of sodium phenylbutyrate and taurursodiol. They countacted Prof Rudy Tanzi – a prominent Alzheimer’s researcher at Harvard – who on a hunch decided to mentor them, and their combined efforts ultimately led to the development of AMX0035. If this drug goes on to ultimately be approved for ALS, this story will become a thing of legend and hopefully encourage other young budding students to take similar bold steps.
The combination of sodium phenylbutyrate and taurursodiol has been found to have a positive impact on survival in a Phase II clinical trial of ALS, providing not only some good news for the ALS community, but also justification for evaluating combination therapies for neurodegenerative conditions. The philosophy in clinical research thus far has always been to identify individual molecules that have a positive effect and then build on them. But that process has been slow and not yielded major results. It has also raised questions, such as what if a specific combination is required for individual molecules to exhibit their effect? (administered by themselves they might not have any effect, but in complement with a second molecule perhaps there is magic). There is a raging debate about this idea, and it will be interesting to see if the CENTAUR study results add fuel to the fire.
Several readers have requested this post, and I hope that they are satified with the outcome. Fingers crossed that we see further positive results in ALS with AMX0035, and perhaps even the initiation of a clinical trial in Parkinson’s.
Stay tuned.
 All of the material on this website is licensed under a
All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
EDITOR’S NOTE – The author of this post is an employee of the Cure Parkinson’s Trust which is supporting the UP study. Neither the UP study team nor CPT has not asked for this post to be written. This post has been provided by the author solely for the purpose of sharing what the author considers very interesting information.
The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by a research scientist, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. Please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from ovrdrv

awesome post, thank you Simon!
We’re currently developing liposomal GM1 for ALS. But the liposome is so far “empty” and could be filled with drugs. I will give it some thought and maybe encapsulate phenylbutyrate and TUDCA as more complex follow-up candidate. GM1, PBA, TUDCA might be synergistic.
LikeLike