|
# # # # At the end of each year, it is a useful process to take stock and review what we have learnt over the last 12 months. 2025 has been a very busy year for Parkinson’s research, with a lot of clinical trial results being reported and new insights being made. In today’s post, we will consider three big Parkinson’s-related research takeaways of 2025 (based on our humble opinions here at the SoPD), and then we will provide an extended overview of some of the important pieces of news from the last 12 months (Be warned: this is a rather long post). # # # # |
 Source: NYTimes
Source: NYTimes
This is little unit is KJ Muldoon.
Research-wise, 2025 was a pretty big year for him (in fact, he was one of Nature’s 10 people who shaped science in 2025).
Born on the 1st August, 2024, in Clifton Heights, Pennsylvania, KJ was the fourth child of Kyle and Nicole Muldoon. The day after he was born it was noted that he was unusually sleepy and averse to feeding. Blood work quickly showed that he was accumulating ammonia in his blood, and a genetic test revealed that he had a mutation that caused carbamoyl phosphate synthetase I (CPS1) deficiency.
KJ was too young for a liver transplant which was the only major treatment available at the time, so his treatment options were looking extremely limited.
What happened next was part of what made 2025 an amazing year:
The kid doesn’t know it yet, but he made medical history as the first human to receive a personalized CRISPR-based gene therapy treatment (Click here to read the research report behind this story). And here is a timeline of events in his treatment story:
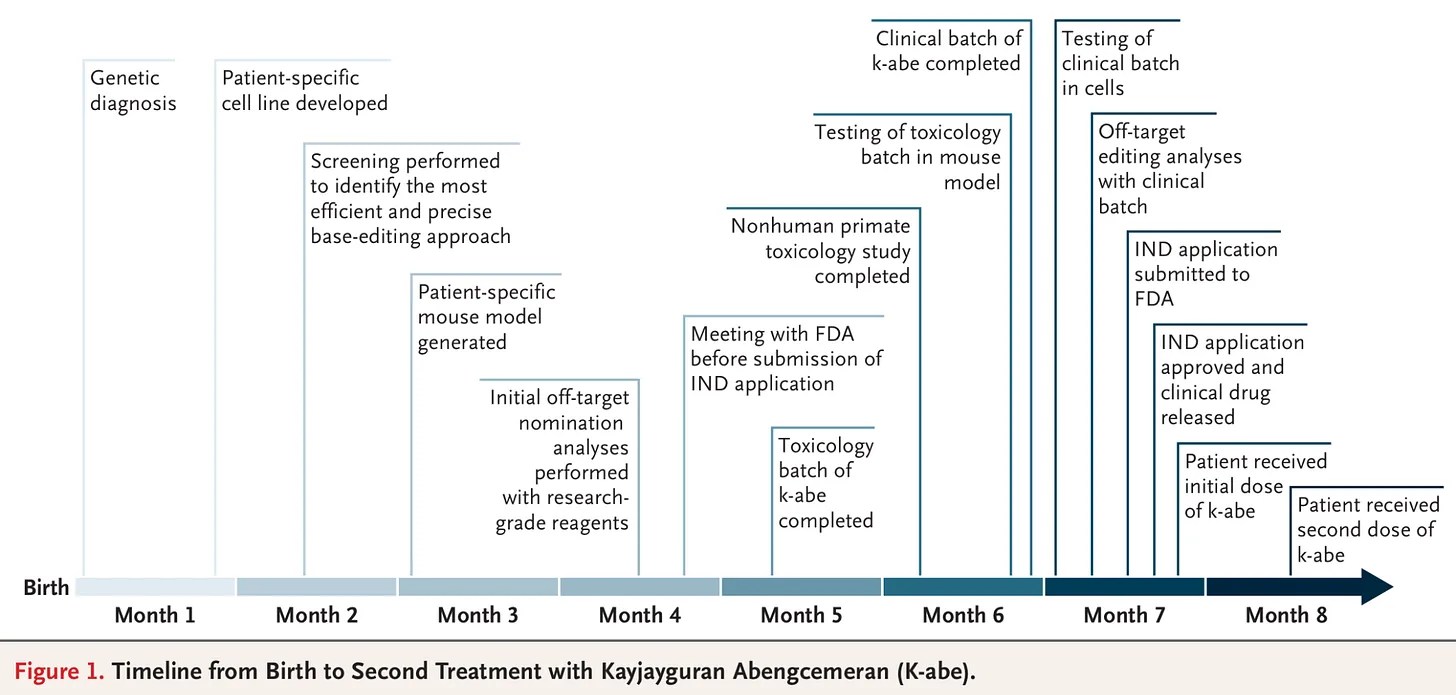 Source: NEJM
Source: NEJM
And very recently, KJ took his first steps (Click here to see this).
Forget about all of the idiotic nonsense flooding social media and all of the witless utterances coming from our so called “leaders” and all of the talking heads on normal media.
Stories like KJ’s are made 2025 an incredible year. Part of humanity truly striving for a better future.
And this was only one story among a huge bag of uncelebrated scientific advances that occurred this year. Advances such as:
- A team of researchers at Roche and Boston’s Children’s Hospital set a new record for the fastest human genome sequencing and analysis. It took them just under 4 hours to sequence the whole genome – in the 2010s it took 3 days (Click here to read more about this).
- The second chikungunya vaccine (‘Vimkunya’) was approved. Chikungunya is a disease spread by mosquitoes (similar to dengue), which can cause months of joint pain and in some rare cases, paralysis. Vaccines do work (Click here to read more about this).
- The seemingly unstoppable growth of renewable energy (Click here to read more about this).
- The FDA gave a green-light to the first multi-person clinical trials of pig kidney transplants, which will hopefully help ease the global shortage of donor organs (Click here to read more about this).
- The Vera C. Rubin Observatory came online.
- The FDA has approved 44 brand new drugs – more than twice as many as were approved in 2010.
- Lenacapavir is a long-acting antiviral injection for HIV received approval in the US for HIV prevention (Click here to read more about this).
- The UK became the first country to offer a vaccine against gonorrhea (Click here to read more about this).
- Researcher combined AI models RFDiffusion and AlphaFold2 to create a ‘multi-step enzyme’ for the first time, and that enzyme has never been seen before in nature (Click here to read more about this and click here to read an exemplar).
- In October, tech company Google announced that its “quantum echoes” algorithm proved 13,000 times faster than a classical computer at predicting molecular structures (Click here to read more about this).
- Mitochondria were discovered to be critical for memory formation in immune T cells and have an unexpected role in tissue healing (Click here and here to read more about this, respectively).
Below is a list of some of the more interesting Parkinson’s research findings of the year – by month, but starting with the top three according to the team here at SoPD HQ.
|
# EDITOR’S NOTE: The author of this blog is the director of research at the medical research charity Cure Parkinson’s. For the purpose of transparency and to eliminate any sense of bias, where Cure Parkinson’s is a funder of the research it shall be noted. The selection of research topics below are based on his opinion alone and do not reflect the thoughts of any other parties. # |
The 3 main SOPD highlights in Parkinson’s-related research for 2025
(in no particular order – just our opinion)