
|
# # # # Tiny genetic variations in a region of DNA called the GBA gene are associated with an increased risk of developing Parkinson’s. The information in the GBA gene provides the instructions for making an enzyme (called GCase) which is involved with waste disposal inside of cells. Individuals with Parkinson’s who carry a variation in their GBA gene typically have low levels of GCase activity, so researchers have been attempting to identify therapeutic molecules that will enhance the level and activity of GCase as an approach towards slowing the progression of Parkinson’s. Recently, however, new research has provide novel insights into how the biology of GCase pathway may be affected in individuals with Parkinson’s who carry a GBA genetic variation. In today’s post, we will explain what the GBA gene and GCase enzyme are, review the new research, and consider the potential implications of these findings. # # # # |
 Prof Sulzer. Source: Youtube
Prof Sulzer. Source: Youtube
Professor David Sulzer is one individual in the scientific research community who truly fascinates me.
In addition to being at the absolute top of his game academically (he is a professor of Psychiatry, Neurology, Pharmacology at Columbia University and maintains a very large research group investigating neurodegenerative conditions), he is also a composer and musician with a discography that any professional artists would be extremely proud of (his recording alias is Dave Soldier).
He’s also written books (for example “Music Math and Mind“).
 Source: Twitter
Source: Twitter
Where he finds the time to do all of these thing I do not know, but I really like the combination of art and science.
Oh, and did I forget to mention the Thai Elephant Orchestra?
I’m sorry: The what?!?
Just watch:
They have released three CDs and the band grew up to 14 elephants.
Fascinating, but what does this have to do with Parkinson’s?
Well, recently Prof Sulzer and colleagues (no elephants included) published a really interesting research report
This is it:
 Title: Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy.
Title: Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy.
Authors: Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, Hutten SJ, Alcalay RN, Kim S, Ximénez-Embún P, Fan L, Kim D, Ko HS, Yacoubian T, Kanter E, Liu L, Tang G, Muñoz J, Sardi SP, Li A, Gan L, Cuervo AM, Sulzer D.
Journal: Sci Adv. 2022 Feb 11;8(6):eabm6393.
PMID: 35138901 (This report is OPEN ACCESS if you would like to read it)
In this report, the researchers wanted to better understand what was happening in cells that carry some of the GBA genetic variants that are associated with Parkinson’s.
What are “the GBA genetic variants”?
The GBA gene is a section of DNA that provides the instructions for making a particular enzyme – that enzyme is glucocerebrosidase (also known as GCase).
 Source: 23andMe
Source: 23andMe
GCase is an enzyme that helps with the digestion and recycling of various types of waste (particularly glucocerebrosides) inside cells (more on that below). Tiny errors (called genetic variants) in the GBA gene are associated with an increased risk of developing Parkinson’s.
They are some of the most common genetic risk factors for the condition.
It is believed that approximately 5%–8% of people with Parkinson’s have a genetic mutation in their GBA gene (Click here and here to read more about this).
How does GCase work and how could this be affecting Parkinson’s?
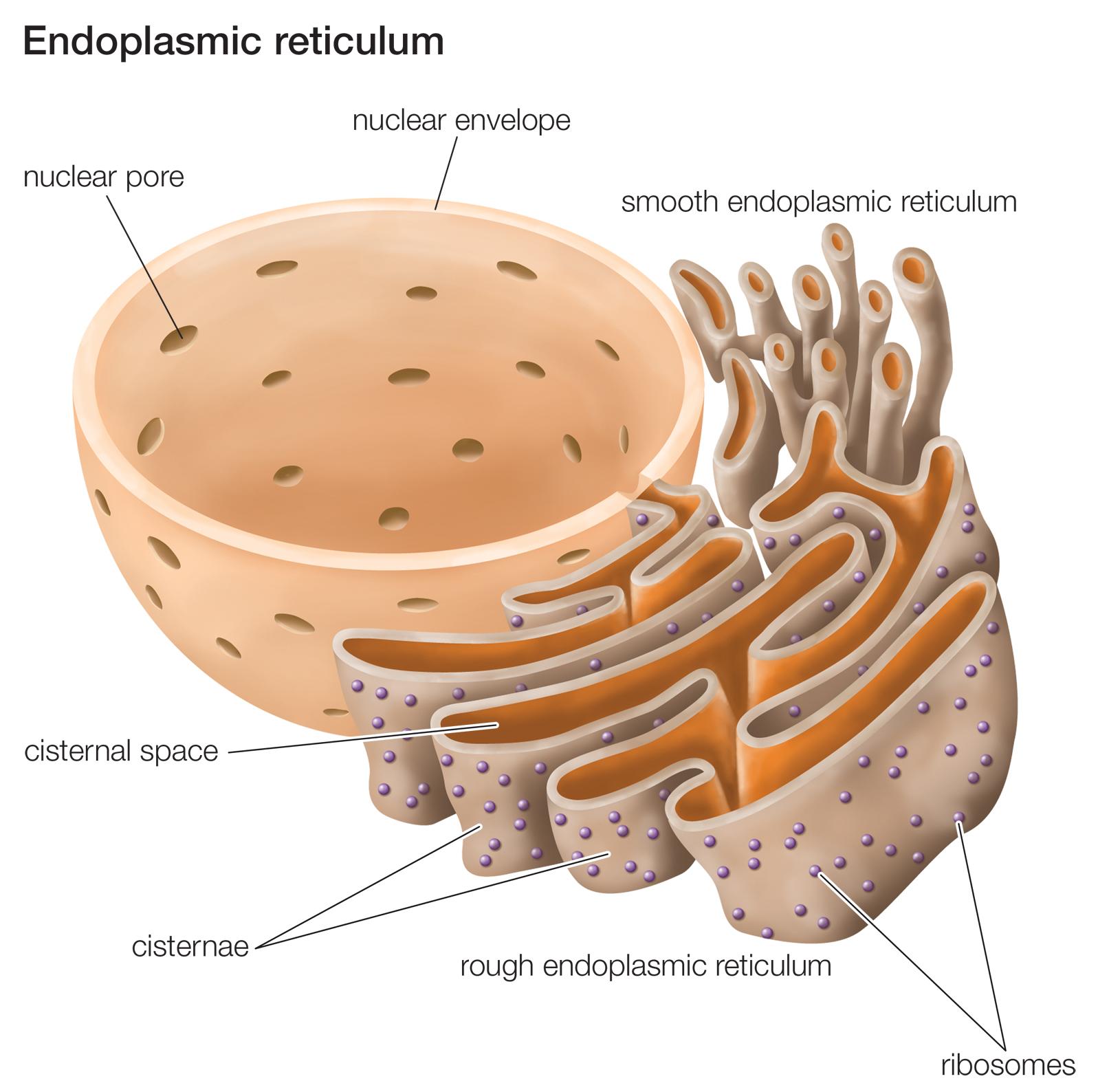
Inside of cells, GCase enzyme is produced in an area called the endoplasmic reticulum.
The endoplasmic reticulum (or ER) is a highly convoluted, netlike mesh structure that extends off the nucleus of a cell. It is the assembly line where proteins are produced within a cell.
 The endoplasmic reticulum. Source: Britannica
The endoplasmic reticulum. Source: Britannica
The nucleus is where the blue prints for making and maintaining an organism is kept in the form of DNA. A template of how to produce a particular protein can be generated from this DNA – that template is called RNA – and that is used to produce a protein (via a process called translation). A large part of that protein production process is conducted within the endoplasmic reticulum.
Once the GCase enzyme is correctly folded, it is transported (by chaperone proteins like LIMP-2) to small bags of digestive enzymes that are called lysosomes, which can be found floating around inside of cells.
 Source: NCBI
Source: NCBI
How do these lysosomes work?
On a relatively continual basis, small parts of a cell membrane are being brought inside the cell. This is a process called endocytosis.
It occurs when the cell needs to consume resources from the outside world in order to find what it requires to function and survive. As a section of cell membrane is brought into the cell, it forms a small spherical bag which is called a vesicle. Given the process by which these outer membrane vesicles are formed, they is referred to as endosomes (sometimes it is also called a vacuole).
 Source: Socratic
Source: Socratic
Once the endosome is inside the cell and detached from the rest of the membrane, it will bind to another vesicle – the lysosome. And as I mentioned above, lysosome is a small bag that is full of digestive enzymes, which help to break down the contents of the endosome.

How lysosomes work. Source: Prezi
The lysosome will fuse with the endosome/vacuole and the enzymes from the lysosome will mix with the material in the vacuole and digest it (or it break down into more manageable components).
This enzymatic process works in a very similar fashion to the commercial products that you use for washing your clothes.
 Enzymatic degradation. Source: Samvirke
Enzymatic degradation. Source: Samvirke
The reagents that you put into the washing machine with your clothes contain a multitude of enzymes, which help to break down the dirty, bacteria, flakes of skin, etc that cling to your clothes. Each enzyme breaks down a particular protein, fat or such like. And this is very similar to the collection of enzymes in the lysosome. All of them are needed to break down all of the contents of the endosome.
GCase is one of these enzymes in the lysosome.
Now, it has previously been assumed that if one of those enzymes – such as GCase – is faulty (due to a genetic mutation), then the enzymatic process will be disrupted and this could result in the build up of un-degraded material over time.
What do you mean by ‘it has previously been assumed’?
Well, there has been a little bit of a mystery associated with GCase research.
Oooh, I like mysteries! What is it?
The thing is, when researchers have looked at the brains of people with Parkinson’s who also carry a GBA genetic variant, they generally do not see a build up of the waste that GCase breaks down.
That waste is primarily glucocerebroside.
In the lysosome, GCase binds to glucocerebroside and chops it up.
Now everyone carries two copies of the each gene in their DNA (one copy from your mother, and the other from your father). Call it mother nature’s insurance policy, but we have two copies of each gene in our DNA. In most cases, if one copy of a particular gene is faulty in an individual, then the other copy can step in and keep things humming along smoothly. But for some genes, this isn’t always the case and GBA seems to be one of those gene (although not everyone with a GBA variant goes on to develop Parkinson’s).
In individuals with Parkinson’s who carry a genetic variant (these cases are referred to as GBA-associated Parkinson’s), they generally have one copy of the GBA gene that produces a normal version of the GCase enzyme. In addition to this, however, they also carry another copy of the GBA gene that has a genetic variation which results in a dysfunctional version of GCase that does not do its job properly. This combination has been associated with reduced levels of GCase activity in cells from people with GBA-associated Parkinson’s.
In the absence of a fully functioning GCase system, one would expect un-digested glucocerebrosides to start piling up over time….
But it doesn’t in the brains of people with GBA-associated Parkinson’s.
This paper here was one of the first to report this finding:
 Title: No evidence for substrate accumulation in Parkinson brains with GBA mutations.
Title: No evidence for substrate accumulation in Parkinson brains with GBA mutations.
Authors: Gegg ME, Sweet L, Wang BH, Shihabuddin LS, Sardi SP, Schapira AH.
Journal: Mov Disord. 2015 Jul;30(8):1085-9.
PMID: 26096906 (This report is OPEN ACCESS if you would like to read it)
In the study, the researchers analysed samples of brain tissue from individuals with GBA-associated Parkinson’s as well as from cases of idiopathic Parkinson’s and compared the results with samples from age matched control brains. While the GBA-associated Parkinson’s cases exhibited 50% reductions in GCase activity, the levels of glucocerebroside were no different between the three groups.
?????
Exactly. It has been a real mystery why we don’t see a build up of undigested glucocerebroside over time.
One that is made more complicated by the fact that other proteins, like Parkinson’s associated protein alpha synuclein do build up in the brains of people with GBA-associated Parkinson’s.
Maybe the low level of GCase enzyme can still manage with the levels of glucocerebroside digestion?
Perhaps. But this only begs the question: why is alpha synuclein accumulating and what is actually causing the neurodegeneration associated with GBA-associated Parkinson’s if it is not a deficiency in GCase activity?
|
# RECAP #1: GCase is an enzyme that is involved with the waste disposal system of cells. It is located in small bags (called lysosomes) of digestive enzymes. People with genetic variants in the region of DNA that provides the instructions for producing GCase have a higher risk of developing Parkinson’s. These individuals have very low levels of GCase activity. # |
So what did Prof Sulzer and his team find?
Prof Sulzer and collaborators wanted to better understand what was happening in cells from people with GBA-associated Parkinson’s. So they grew cells in culture that carried different GBA genetic variants and compared their activity with normal/unaffected cells.
They found that in the normal cells, the GCase enzyme was efficiently being transported to the lysosomes. But the cells that carried GBA variants had very low levels of GCase inside their lysosomes. They had high levels of the enzyme in the endoplasmic reticulum (the assembly line of cellular proteins), but it didn’t appear to be transported to the lysosome.
To be sure about this, the researchers made a close inspection of the lysosomes in these cells, and when they did they made an interesting discovery: In the cells carrying GBA variants, they found some of the dysfunctional form of the GCase enzyme stuck on the outside wall of the lysosomes.
They reported that a portion of the dysfunctional form of the GCase enzyme was being recognized by some of the chaperones that transport it to the lysosome. Not only this, but some of the dysfunctional form of the GCase enzyme was actually being transported to the outer surface of the lysosome.
This is a good thing right?
It’s more complicated than that.
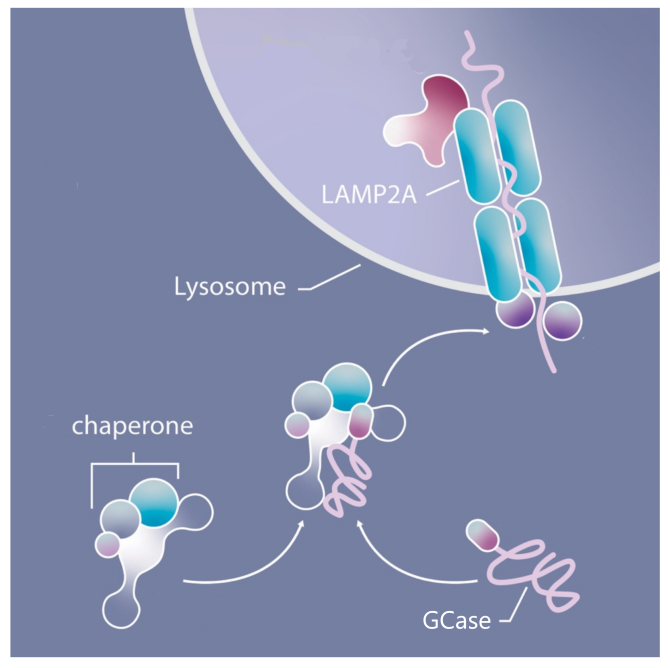
Once it arrives at the lysosome, GCase interacts with a protein called LAMP2A.
 Adapted from source: MDPI
Adapted from source: MDPI
Lysosome-associated membrane protein type 2A (or LAMP2A) is a receptor on the surface of lysosomes that is involved with transferring molecules like GCase into the interior of the lysosome. It helps to get GCase into the lysosome where it can become active and do its job.
But Prof Sulzer and colleagues found that when the dysfunctional form of the GCase enzyme arrived at the lysosome and interacted with LAMP2A, it disrupted the normal activity of LAMP2A. This resulted in an accumulation of the dysfunctional form of the GCase enzyme on the outer (or cytosolic) surface of the lysosomes.
By interacting with LAMP2A and inhibiting its function, the dysfunctional form of the GCase enzyme also bunged up the works for the transfer of the normal form of GCase enzyme into the lysosome.
“The normal form of GCase enzyme”?
Remember that people with GBA-associated Parkinson’s have two copies of the GBA gene (and thus two versions of GCase enzyme being produced in cells): the one carrying the genetic variation (resulting in a dysfunctional form of the GCase enzyme) and a normal, functional form of the GCase enzyme. This normal form should be able to still do its function, but according to this new research the dysfunctional form of the GCase enzyme is inhibiting LAMP2A and blocking the normal form of GCase from being transferred into the lysosome.
The result: An accumulation of GCase enzyme (both versions) on the outside (or cytosolic) surface of the lysosomes.
The researchers reported that “Approximately half of the GCase in PD-GBA subjects was present on the cytosolic side of the lysosomes, whereas no detectable GCase was present on the cytosolic side of lysosomes from healthy controls”
Does that build up of dysfunctional GCase cause problems for any other proteins trying to enter the lysosome?
The researchers were wondering the same thing. They next hypothesized that this accumulation of the dysfunctional form of the GCase enzyme and inhibition of LAMP2A on the outer lysosomal membrane might disrupt the transfer of other proteins into the lysosome.
And yep, they were right.
They found that it lead to accumulation of the Parkinson’s-associated protein alpha synuclein inside of cells.
You see, LAMP2A is required for the shifting of alpha synuclein into the lysosome for degradation, so anything that blocks LAMP2A from doing its job results in increased levels of alpha synuclein. Artificially increasing levels of LAMP2A has been shown to rescue cells from alpha synuclein-associated toxicity (Click here to read more about this).
Prof Sulzer’s team also reported a similar build up of another protein associated with neurodegenerative conditions – Tau – with the accumulation of the dysfunctional form of the GCase enzyme on the outer lysosomal membrane (Click here to read an old SoPD post about Tau).
So the reduced levels of GCase activity were not influential in this situation?
The researchers double checked all of their results with further experiments and they found that the disruptive effect of the dysfunctional form of the GCase enzyme on lysosomal function was not due to loss of GCase enzymatic activity inside of lysosomes. They conducted two sets of experiments to confirm this. One involved treating cells with a GCase inhibitor, while the other involved cells that do not produce any GCase, and neither situation resulted in any decreased levels of waste disposal.
Prof Sulzer and colleagues next conducted experiments investigating if this LAMP2A inhibitory effect of the dysfunctional form of the GCase enzyme could influence the neurodegeneration associated with GBA-associated Parkinson’s. They found that it could, but the effect required the presence of alpha synuclein, as dopamine neurons with no alpha synuclein did not die in these studies.
Overall, the study demonstrated a new potential mechanism of neurodegeneration (which needs to be independently replicated) and reinforced the importance of not simply ignoring the mutant version of particular protein. It might suggests that simply raising GCase levels may not be enough for correcting GBA-associated Parkinson’s.
|
# # RECAP #2: A new study has found that a small fraction of the dysfunctional form of the GCase enzyme (resulting from GBA genetic variants associated with Parkinson’s) may be transported to the surface of the lysosome, but it can not enter the structure. Instead, it accumulates and blocks the entrance mechanism. This has a flow on effect, resulting in the blocking of other proteins from entering the lysosome, including the neurodegeneration-associated proteins alpha synuclein and Tau. This mechanism appears to result in cell death. # # |
So what does this mean for efforts to therapeutically increase levels of GCase activity?
This is a really good question.
One which was not addressed in the report and probably depends on the nature of each experimental therapy. But it would be very interesting to test some of these agents within this new paradigm (if the results are independently replicated).
But the question is particularly important to yours truly and one particular experimental agent.
Full disclosure: The author of this blog is an employee of Cure Parkinson’s, which is an international funder of research focused on slowing/stopping/reversing Parkinson’s.
![]()
And the charity has been supporting the clinical repurposing of the cough therapy ambroxol for Parkinson’s (Click here to read a previous post about ambroxol).
 Source: Skinflint
Source: Skinflint
So the first question that passed through my head when I read the report from Prof Sulzer’s team was: “what does ambroxol do in this situation?”
Remarkably, part of the answer to this question came in the form of a report published just a few days after the Sulzer paper.
This is the report here:
 Title: Ambroxol reverses tau and α-synuclein accumulation in a cholinergic N370S GBA1 mutation model.
Title: Ambroxol reverses tau and α-synuclein accumulation in a cholinergic N370S GBA1 mutation model.
Authors: Yang SY, Taanman JW, Gegg M, Schapira AHV.
Journal: Hum Mol Genet. 2022 Feb 18:ddac038. Online ahead of print.
PMID: 35179198
In this study, the researchers grew cholinergic neurons in cell culture. Some of these cells carried Parkinson’s-associated GBA genetic variants (similar to the ones investigated in Prof Sulzer’s report).
What are cholinergic neurons?
Cholinergic neurons are cells in the brain that produce and send message using a neurotransmitter called acetylcholine.
While dopamine neurons are associated with movement, cholinergic neurons are generally more involved with cognitive ability. And the researchers behind this study wanted to know what impact Parkinson’s-associated GBA genetic variants could have on the functioning of cholinergic neurons.
Importantly, while they were conducting their investigations, they were also testing ambroxol to determine if it could rescue any observed issues.
The researchers reported significant reductions in levels of GCase enzyme and activity in the cholinergic neurons carrying Parkinson’s-associated GBA genetic variants. And levels of both tau and alpha synuclein protein were significantly increased in these cells.
Crucially, they found that ambroxol treatment significantly enhanced GCase activity in these cells, and decreased levels of both tau and α-synuclein.
(Reading this, I breathed a big sigh of relief!)
That’s in cholinergic neurons. What about other types of cells?
Back in 2014, the same research team published this report addressing this question:

Title: Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells.
Authors: McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, Schapira AH.
Journal: Brain. 2014 May;137(Pt 5):1481-95.
PMID: 24574503 (This report is OPEN ACCESS if you want to read it)
In this study, the investigators collected skin cells (called fibroblasts) from eleven people with GBA variants (some of whom had been diagnosed with Parkinson’s). They measured the amount of GCase protein and enzyme activity in these cells, and they found that GCase enzyme activity was significantly reduced in fibroblasts from GBA mutations compared to normal fibroblasts (on average the enzyme was acting at just 5% of normal levels in the cells with GBA variants).
They found that ambroxol treatment increased GCase activity in fibroblasts from people with GBA variants and in fibroblasts from healthy controls. Ambroxol treatment also reduced markers of oxidative stress and levels of alpha-synuclein in the GBA variant carrying cells.
And importantly, these results have been replicated by other independent research groups (Click here and here to read examples).
Collectively, these results suggest that ambroxol is still worthy of clinical testing in individuals with GBA-associated Parkinson’s.
(Another big sigh of relief)
So what does it all mean?
Researchers have presented new data indicating a novel mechanism by which genetic variants associated with Parkinson’s could be playing a role in the cell death associated with the condition. The mechanism demonstrates that the dysfunction form of the protein (caused by the genetic variant) does matter and may need to be addressed in order to correct the course of the condition.
The next step (once this new paradigm has been independently replicated) will be to test some of the agents currently being experimentally tested as therapeutic interventions. Such experiments may provide further insights into the biology underlying the condition, and provide greater justification for their clinical testing.
 All of the material on this website is licensed under a
All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
Further, the author of this post is an employee of the charitable research trust Cure Parkinson’s. The Trust has not asked for this post to be written. This post has been written by the author solely for the purpose of sharing what the author considers interesting information.
The banner for today’s post was sourced from medicalnewstoday

simon
most interesting; now that i have a diagnosis of PSP which as you will know is predominantly involving the TAU protein. Is this exclusive or does the alpha synuclein play a part too?
kind regards
Keith
LikeLike
I have been on Ambroxol for two years and have not progressed at all.
LikeLike
Does ambroxol help those with lewy body as well as it does with thos who have parkinson’s only?
LikeLike
“Where he finds the time to do all of these thing I do not know…” said the guy who does this blog. 🙂
LikeLike
https://pubmed.ncbi.nlm.nih.gov/30693689/
“curcumin…increased protein expression of…LAMP2A. When curcumin and 3-MA [an autophagy inhibitor–LT] were given concurrently, the number of surviving dopamine neurons, [and] protein expression of…LAMP2A…increased (P<0.05 or P<0.01), and both protein expression and mRNA expression of α-Syn decreased (P<0.05 or P<0.01) compared with MPTP model group; but the number of surviving dopamine neurons and protein expression of LAMP2A and LC3-Ⅱ decreased compared with curcumin group (all P<0.05)."
The above seems to indicate that curcumin provides an enhanced number of LAMP2A receptors on the surface of lysosomes, which might add additional LAMP2A receptors beyond those disabled by dysfunctional (mutated) copies of GCase, allowing the normal copies of GCase (e.g., from the parent who did not have the GBA mutation) additional opportunities to enter the lysosomes. And also, allowing those additional unmolested (by mutated GCase) LAMP2A receptors to shepherd alpha synuclein into the lysosomes for disposal.
Might this mean that curcumin could be either an alternative or an adjunctive therapy to Ambroxol?
LikeLike