|
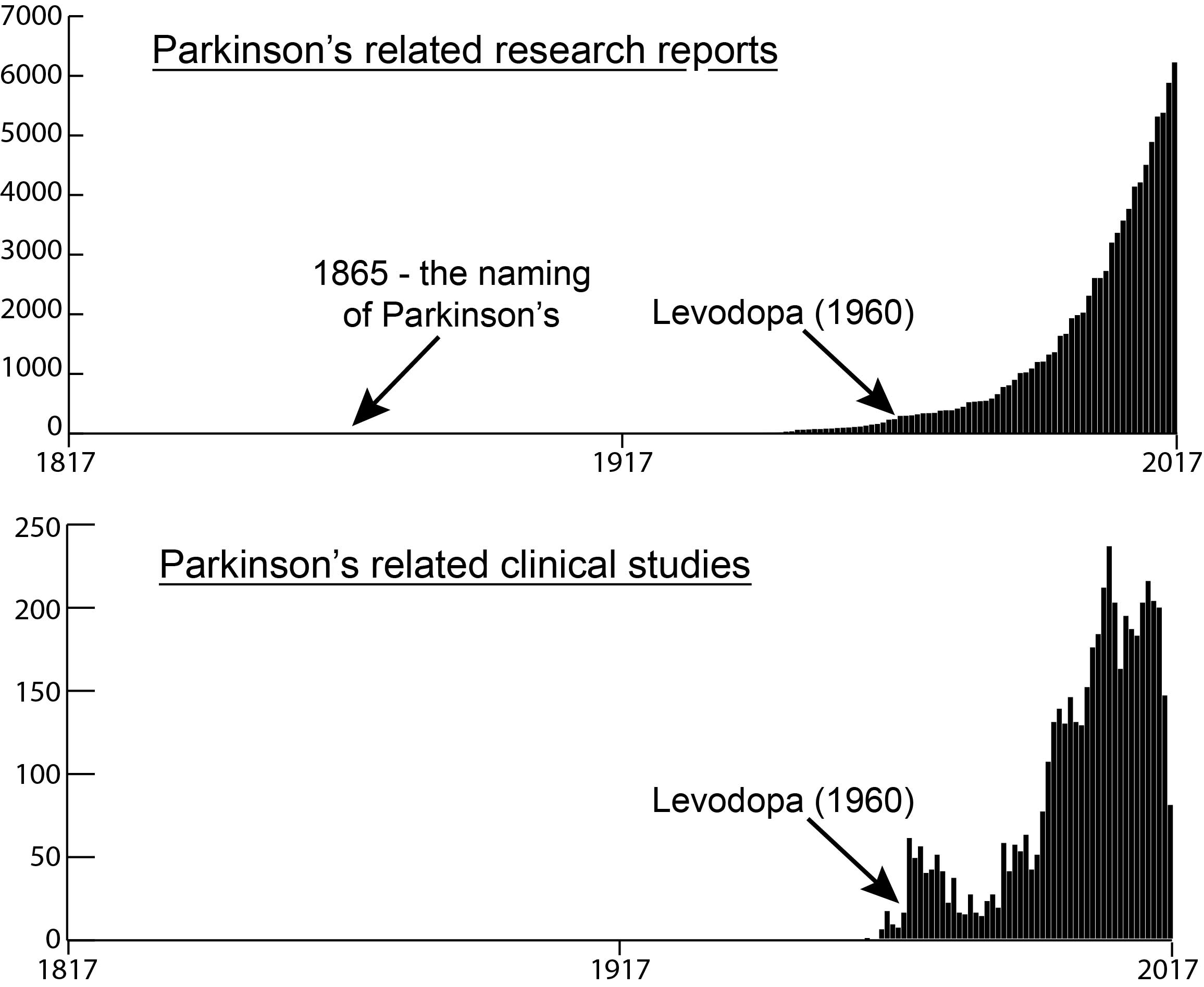
# # # # This week an announcement was made regarding The Edmond J. Safra Accelerating Clinical Treatments for Parkinson’s Disease (EJS-ACT PD) Initiative. It is hoping to revolutionise the way clinical trials for potentially disease-modifying drugs for Parkinson’s are conducted. The project is focused on the setting up a multi-arm, multi-stage (MAMS) platform for evaluating new therapies for PD. In today’s post, we will discuss what MAMS trials involve and the current details of the EJS-ACT PD initiative. # # # # |
 Source: Motionarray
Source: Motionarray
This week I boarded a train for the first time in 16 months and made my way down to London. It felt a wee bit surreal.
I arrived at Liverpool street station and was immediately shocked by the lack of crowds, the lack of face masks (seriously?!? I’ve had my two jabs as well, but I’m still wearing my mask – you are nuts if you don’t!), and the large number of empty shops. How the world has changed.
In the early morning light, I walked across central London towards St Pancras station – the weather was spectacular and it was an incredible pleasure to stroll through some old stomping grounds.
 Source: Parksandgardens
Source: Parksandgardens
At St Pancras station, I made my way to the enormous Francis Crick institute, where a group of Parkinson’s researchers and advocates were gathering for a really intriguing meeting.
 Source: Timeshighereducation
Source: Timeshighereducation
What was the meeting about?