
|
An Advanced Glycation Endproduct (or AGE) is a protein or lipid that has become glycated. Glycation is a haphazard process that impairs the normal functioning of molecules. It occurs as a result of exposure to high amounts of sugar. These AGEs are present at above average levels in people with diabetes and various ageing-related disorders, including neurodegenerative conditions. AGEs have been shown to trigger signalling pathways within cells that are associated with both oxidative stress and inflammation, but also cell death. RAGE (or receptor of AGEs) is a molecule in a cell membrane that becomes activated when it interacts with various AGEs. And this interaction mediates AGE-associated toxicity issues. Recently researchers found that that neurons carrying the Parkinson’s associated LRRK2 G2019S genetic variant are more sensitive to AGEs than neurons without the genetic variant. In today’s post we will look at what AGE and RAGE are, review the new LRRK2 research, and discuss how blocking RAGE could represent a future therapeutic approach for treating Parkinson’s. |
The wonder of ageing. Source: Club-cleo
NOTE: Be warned, the reading of this post may get a bit confusing. We are going to be discussing ageing (as in the body getting old) as well as AGEing (the haphazard process processing of glycation). For better clarification, lower caps ‘age’ will refer to getting old, while capitalised ‘AGE’ will deal with that glycation process. I hope this helps.
Ageing means different things to different people.
For some people ageing means more years to add to your life and less activity. For others it means more medication and less hair. More wrinkles and less independence; more arthritis and less dignity; More candles, and less respect from that unruly younger generation; More… what’s that word I’m thinking of? (forgetfulness)… and what were we actually talking about?
Wisdom is supposed to come with age, but as the comedian/entertainer George Carlin once said “Age is a hell of a price to pay for wisdom”. I have to say though, that if I had ever met Mr Carlin, I would have suggested to him that I’m feeling rather ripped off!

George Carlin. Source: Thethornycroftdiatribe
Whether we like it or not, from the moment you are born, ageing is an inevitable part of our life. But this has not stopped some adventurous scientific souls from trying to understand the process, and even try to alter it in an attempt to help humans live longer.
Regardless of whether you agree with the idea of humans living longer than their specified use-by-date, some of this ageing-related research could have tremendous benefits for neurodegenerative conditions, like Parkinson’s.
What do we know about the biology of ageing?
In 2013, a very detailed review was written on the topic of the biology of ageing, in which the authors proposed nine molecular “hallmarks” of ageing:

Title: The hallmarks of aging
Authors: López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G.
Journal: Cell. 2013 Jun 6;153(6):1194-217.
PMID: 23746838 (This article is OPEN ACCESS if you would like to read it)
In this review, the authors outlined 9 biological aspects of ageing and reviewed 3 biological pathways that can influence the ageing process. The 9 biological aspects of the body getting old are:
- Genomic instability – the longer we live, the more errors (or mutations/variations) we accumulate in our the DNA. DNA provides the blueprint or the instructions for making and maintaining a normal cell. To complicate this issue, we have multiple forms of DNA within each of our cells: there is DNA in the nucleus of each cell of our body, but then there is also the DNA within each mitochondria in each cell of our body. Mitochondria are the power stations of our cells and they have their own DNA. Each time a cell or a mitochondria divides, there is the opportunity for the replication to have tiny alterations, and over the course of an entire life there is a lot of cell division. But even within cells that don’t divide (such as neurons in the brain) there is also the chance for mutations in the DNA to occur (via exposure to toxins, radiation, etc). We looked at this phenomenon in the previous post – Click here to see that post).

DNA damage. Source: Eyeonicr
- Telomere attrition – Telomeres are the terminal caps on the end of chromosomes (the tightly bound structures of DNA in your cells). They are believed to protect the DNA at the end of the chromosomes from being damaged. With each cell division and as individuals age, these telomeres get a wee bit shorter. They can be replenished by an enzyme called telomerase reverse transcriptase, but most mammalian cells do not produce this enzyme. The average telomere length has thus become viewed as a ‘replicative clock’ within populations of cells, and ‘telomere attrition’ is associated with cells becoming senescent (they stop dividing). When mice are genetically engineered to produce a lot of telomerase reverse transcriptase, they have a delay in ageing accompanied by an “extension of the median life span” (Click here to read about this research). Would you like to live forever? There are researchers targeting telomerase for both the treatment of cancer and longevity (Click here to read more about this).

Source: Nestacertified
- Epigenetic alterations – ‘Epigenetics’ is anything other than DNA sequence that influences the development/maintenance of an organism. And these processes plays a major role in ageing. DNA methylation is a good example of epigenetics. DNA methylation is a process by which methyl groups are added to DNA. A methyl group is an alkyl derived from the chemical methane. When methyl groups are added to DNA, it blocks the DNA from being transcribed (the first step in the production of protein). The gene that has been methylated is essentially shut down. And many of the genes that are important in the early years of life are methylated during the later years of life. Cells from people with progeria (a rare genetic condition that make affected individuals appear to be older than they are) exhibit DNA methylation patterns that are very similar to those found in normal ageing. Sam Berns (the young man in the video below) was diagnosed with Progeria at 2 years of age – his outlook on the world is pretty cool:
- Loss of proteostasis – Loss of protein homeostasis (proteostasis) is a very common feature of the ageing process and it is particularly relevant in neurodegenerative conditions like Parkinson’s which are characterised by the appearance of ‘non-native’ (or mis-folded) proteins – Click here for a good review on this topic. Proteostasis involves mechanisms for the stabilising correctly folded proteins, correcting mis-folded proteins, and disposing of old or mis-folded proteins. Many studies have now demonstrated that proteostasis is altered with the ageing process (Click here for a good review on this topic). And there have been studies suggesting that improving proteostasis can delay ageing in mammals (Click here to read an example of this).

Proteostasis. Source: Ncbi
- Deregulated nutrient sensing – It might sound silly, but metabolic activities inside a cell can actually put a great deal of stress on that cell. Too much activity and too many changes in nutrient availability/composition causes cells to age faster. The byproducts (or waste) of this process build up over time, which results in damage to cells (via oxidative stress, mitochondrial dysfunction, etc). To counter this, organisms have developed multiple nutrient sensing pathways to make sure that the body takes in just the right amount of nutrition – not too much, not too little: “Goldilocks”. As we age, however, there is a deregulate the nutrient-sensing molecules and downstream pathways. Age-related obesity, diabetes and other metabolic syndromes result. Consistent with this idea, dietary restriction has been shown to increase lifespan in all investigated species investigated, from unicellular organisms to primates (Click here for a good review on the science of dietary restriction). Whether this approach is suitable for Parkinson’s (where lower body weight is associated with poorer outcomes) is yet to be determined.
- Mitochondrial dysfunction – We have discussed mitochondria many times on this website. They are the power stations of a cell, providing the cell with energy in exchange for room and board. And as cells age, the production of that energy is reduced. And there is a lot of evidence to suggest that mitochondrial dysfunction can accelerate ageing in mammals (Click here, here and here for examples of studies supporting this idea).

Mitochondria and their location in the cell. Source: NCBI
- Cellular senescence – This is the phenomenon by which normal cells cease to divide. It is generally considered an anti-cancer mechanism that occurs in cells which are capable of division. Think of senescence as a handbrake that is pulled inside of a cell to stop it from having the ability to divide. But the problem is that after this “brake” has been pulled, these senescent cells remain in the body and start to accumulate with age. These cells are still active, but functionally altered, which has been shown to have negative consequences (we recently reviewed new research on this topic with regards to Parkinson’s – click here to read that post).
- Stem cell exhaustion – Within our bodies there are dividing cells, which are referred to as stem cells. As we age, these cells slow down their rate of division and eventually begin to lose their ability to divide. For example, hematopoiesis (the production of blood cells from stem cells in bone marrow) declines with ageing, resulting in a reduced production of immune cells (a process termed immunosenescence – Click here to read more about this). We also see this reduction in stem cells in the brain (click here to read more about this) and in muscle (click here for more information on this). A reduced pool of stem cells is obviously going to have a detrimental impact on the long-term maintenance of an organism.

Source: Eduboard
- Altered intercellular communication – Beyond changes within cells, ageing is also associated with changes in intercellular communication. Cells must be able to communicate between each other for the body to function properly. As we age, this system of communication becomes reduced and this can have very negative results for the body. Inflammation (or inflammaging) is an example of this: the accumulation of pro-inflammatory can damage tissue, the failure of a reduced immune system to properly remove pathogens and dysfunctional cells, accumulating senescent cells secreting pro-inflammatory molecules, etc. There is very interesting, proof-of-principle data that suggests the possibility for rejuvenation via blood-borne systemic factors (Click here to read more about this).
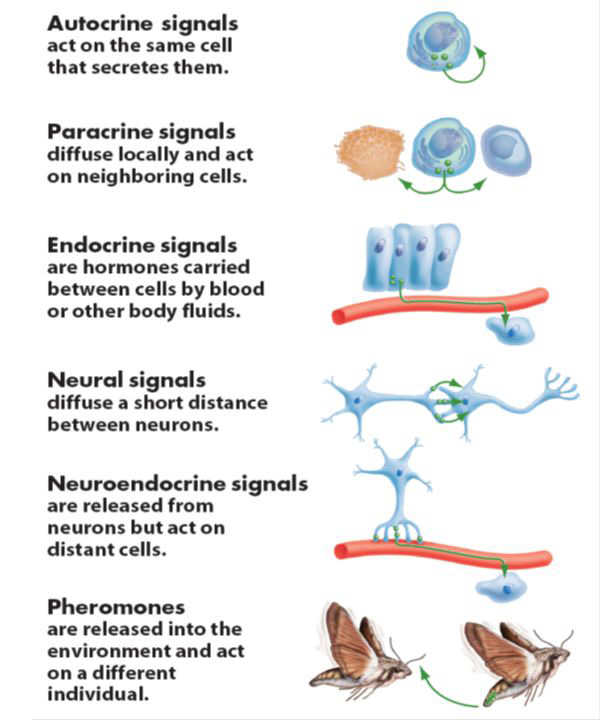
Different methods of cellular communication. Source: Slideplayer
Readers of this blog may be thinking to themselves that many of these aspects of ageing are also known to play a role in the development of Parkinson’s (such as the loss of proteostasis), and this has led some to question whether Parkinson’s may simply be a condition that results from ageing (Click here to read more about this). The jury, however, is still out on this idea.
While this review provides an excellent overview, the list of biological features of ageing is not exhaustive.
For example, one aspect of ageing that this review article did not address was the gradual build up of AGEs.
Ok, so that’s the capitalised version of ‘AGE’, can you remind me again what it means?
As we said above, an Advanced Glycation End product (AGE) is a protein or lipid that has become glycated. This video will give you the basics:
Glycation is the non-enzymatic reaction between reducing sugars (such as glucose) and proteins or nuclei acids (think DNA/RNA). Glycation should be distinguished from glycosylation, which is an enzyme-based reaction between sugars and proteins that approximately half of all proteins in a cell undergo.
Unlike glycosylation, Glycation is a haphazard process. And this uncontrolled activity has the effect of impairing the normal functioning of molecules.
Glycation is known to affect the conformation and function of proteins such as hemoglobin (Hemoglobin is the protein molecule in red blood cells that carries oxygen from the lungs to the body’s tissues – Click here to read more about the glycation of this protein) and albumin (albumin is a protein made by the liver that plays an important in regulating blood volume – Click here to read more about the glycation of this protein).
It generally occurs as a result of exposure to high amounts of sugar.

Source: Slowaging
AGEs affect nearly every type of cell and molecule in the body, and they are believed to play a causative role in the vascular complications of diabetes.
Critically, AGE formation is irreversible.
Are there any connections between high sugar levels and Parkinson’s?
Sort of.
In 1974, a small study was published in the Journal of Chronic Diseases that presented a rather startling set of results:

Title: Glucose intolerance in Parkinson’s disease.
Authors: Lipman IJ, Boykin ME, Flora RE.
Journal: J Chronic Dis. 1974 Dec;27(11-12):573-9.
PMID: 4436423
In the study, Lipman and colleagues conducted some routine glucose tolerance tests on a group of 56 people with Parkinson’s (7 additional subjects with Parkinson’s were excluded because they had been previously diagnosed with diabetes).
After being asked to fast overnight, the subjects were then given 100g of glucose and blood samples were collected from them every hour for 3 hours. When the glucose levels in the blood were measured and compared with the results of 5 previous studies conducted on normal healthy adults of the same age (one of those studies involved 7000 participants), it was found that the people with Parkinson’s in the Lipman study had a much higher average level of glucose in their blood than all of the other 5 studies looking at healthy individuals.
Shockingly, almost half (46.4%) of the participants in the Lipman study actually fulfilled the criteria for a diagnosis of diabetes.
More recent survey data has revealed that diabetes is established in between 8–30% of people with Parkinson’s (click here for more on this) – obviously this is in excess of the approximately 6% prevalence rate in the general public (Source: DiabetesUK).
What is diabetes?
‘Diabetes mellitus’ is what we commonly refer to as diabetes. It is basically a group of metabolic conditions that share a common feature: high blood sugar (glucose) levels for a prolonged period.

There are three types of diabetes:
- Type 1, which involves the pancreas being unable to generate enough insulin. This is usually an early onset condition (during childhood) and is controlled with daily injections of insulin.
- Type 2, which begins with cells failing to respond to insulin. This is a late/adult onset version of diabetes that is caused by excess weight and lack of exercise.
- Type 3, occurs during 2-10% of all pregnancies, and is transient except in 5-10% of cases.
What is this stuff called insulin?
Insulin is a hormone – that our body makes – which allows us to use sugar (glucose) from the food that you eat. Glucose is a great source of energy. After eating, our body is releases insulin which then attaches to cells and signals to those cells to absorb the sugar from our bloodstream. Without insulin, our cells have a hard time absorbing glucose. Think of insulin as a “key” which unlocks cells to allow sugar to enter the cell.
Ok, so how is it all connected to Parkinson’s?
The short answer is ‘we currently don’t know’.
There have, however, been numerous studies now that suggest an association between diabetes and Parkinson’s. The first of these studies was:

Title: Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease.
Authors: Driver JA, Smith A, Buring JE, Gaziano JM, Kurth T, Logroscino G.
Journal: Diabetes Care. 2008 Oct;31(10):2003-5.
PMID: 18599528
In this study, 21,841 male doctors (participants in the Physicians’ Health Study) were followed over 23 years. The researchers found that people with diabetes had an increased risk of developing Parkinson’s risk. Interestingly they reported that the highest Parkinson’s risk was seen in individuals with short-duration, older-onset diabetes.
In another study:

Title: Diabetes and risk of Parkinson’s disease.
Authors: Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, Chen H.
Journal: Diabetes Care. 2011 Apr;34(4):910-5. doi: 10.2337/dc10-1922. Epub 2011 Mar 4.
PMID: 21378214
This study came from another long term study, which was following 288,662 participants of the National Institutes of Health-AARP Diet and Health Study. The researchers found that the risk of Parkinson’s was approximately 40% higher among diabetic patients than among participants without diabetes. In this study, however, the analysis showed that the risk was largely limited to individuals who had diabetes for more than 10 years.
A third study:

Title: Diabetes and the risk of developing Parkinson’s disease in Denmark.
Authors: Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B.
Title: Diabetes Care. 2011 May;34(5):1102-8.
PMID: 21411503
Using data from the nationwide Danish Hospital Register hospital records, the researchers found that having diabetes was associated with a 36% increased risk of developing Parkinson’s. Interestingly, they reported that the risk was stronger in women and people with early-onset Parkinson’s (eg. diagnosed before the age of 60 years).
A recent meta-analysis of 7 population-based cohort studies also suggested that diabetes is associated with a 38% increased risk of developing Parkinson’s (Click here to read more about this research).
For a really good recent review on diabetes, AGEs and Parkinson’s, I can recommend a review article from the Journal of Parkinson’s – Click here to read the article.
Interesting. But what does glycation and AGE have to do with Parkinson’s? Has anyone every looked at AGEs in Parkinson’s?
So way back in the year 2000, this research report was published:

Title: Crosslinking of alpha-synuclein by advanced glycation endproducts–an early pathophysiologicalstep in Lewy body formation?
Authors: Münch G, Lüth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P.
Journal: J Chem Neuroanat. 2000 Dec;20(3-4):253-7.
PMID: 11207423
In this study, the researchers observed that AGEs and the Parkinson’s associated protein alpha synuclein were similarly distributed in very early Lewy bodies in the human brain. Lewy bodies are dense clusters of protein that are present in the brains of people with Parkinson’s.
In this study, however, the sections of postmortem brain were from cases that had no previous history of obvious neurological issues (no Parkinson’s). All of these cases though, exhibited some Lewy bodies. The investigators considered these cases “pre-Parkinson patients” – in other words, the person passed away before developing the clinical signs of Parkinson’s (and yes, I appreciate that this is a bit of a stretch).
In the substantia nigra – the region where the dopamine neurons reside, and an area of the brain affected by Parkinson’s – the researchers found that among 400–600 dopamine neurons, one to two neurons showed the presence of Lewy bodies. All of the Lewy bodies had high levels of the Parkinson’s-associated protein, alpha synuclein, AND all of the Lewy bodies exhibited the presence of AGEs. Similar to alpha synuclein, the presence of AGEs was most dense in the outer zone of the Lewy bodies. In the image below you can see a circular Lewy body in the middle of both panels (F & F’). The green is labelling AGEs, while the red coloration highlights the location of alpha synuclein:

AGEs in Green & alpha synuclein in Red. Source: Sciencedirect
The presence of AGEs in Lewy bodies in non-Parkinsonian brains suggested to the researchers that AGEs may be involved in promoting the formation of Lewy bodies in Parkinson’s as well, and their results were actually supported by some previous research results:

Title: Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease.
Authors: Castellani R, Smith MA, Richey PL, Perry G.
Journal: Brain Res. 1996 Oct 21;737(1-2):195-200.
PMID: 8930366
In this study, the researchers looked at Lewy bodies on sections of brain from four postmortem cases of Parkinson’s and three cases of Lewy body dementia. They found AGEs present in almost all of the Lewy bodies, particularly in the midbrain and brain stem regions. And this research has been further supported by a more recent research report that looked Lewy bodies in cases of incidental Parkinson’s:

Title: Evidence of oxidative stress in the neocortex in incidental Lewy body disease.
Authors: Dalfó E, Portero-Otín M, Ayala V, Martínez A, Pamplona R, Ferrer I.
Journal: J Neuropathol Exp Neurol. 2005 Sep;64(9):816-30.
PMID: 16141792
In this study, the researchers undertook an analysis of AGEs in the frontal cortex, amygdala, and substantia nigra (the region of the brain where the dopamine neurons reside – those same cells which are lost in Parkinson’s) in postmortem cases where there was no reported neurologic issues, but also cases where incidental Parkinson’s had been pathologically verified.
All of these studies indicate that AGEs are present in Lewy bodies. But the AGEs and Parkinson’s research does not stop with just the presence of AGEs in Lewy bodies:

Title: Advanced glycation end products induce in vitro cross-linking of alpha-synuclein and accelerate the process of intracellular inclusion body formation.
Authors: Shaikh S, Nicholson LF.
Journal: J Neurosci Res. 2008 Jul;86(9):2071-82.
PMID: 18335520
In this study, the (Kiwi!) researchers found that exposing the human alpha synuclein protein to AGEs for different periods of time promoted the cross-linking of alpha synuclein proteins in cell culture. This means that the AGEs were helping in some way to bind the individuals alpha synuclein proteins together – similar to what we see in the situation of Parkinson’s perhaps. The investigators thought that this was rather interesting and decided to look deeper into what was actually happening.
Next, they treated cells with a neurotoxin that is associated with Parkinson’s (a pesticide called Rotenone). As expected, Rotenone caused oxidative damage and mitochondrial problems within the cells, but the researchers were more interested to see what happened to AGEs and alpha synuclein. They noticed the formation and intra-cellular (inside the cell) accumulation of AGEs followed the oxidative damage and mitochondrial events. Importantly, the appearance of the AGEs preceded the build-up of alpha synuclein within the cell – that is to say, the AGEs came before the alpha synuclein protein (in this ‘egg before the chicken’-like situation).
They also reported that high levels of extra-cellular (outside the cell) AGEs could accelerate the formation of intra-cellular AGEs which also preceded the accumluation of alpha synuclein.
Interesting huh?
But wait: there’s more (and this is where the RAGE part comes into the story!)
Last year, this research report was published:

Title: Targeted inhibition of RAGE in substantia nigra of rats blocks 6-OHDA-induced dopaminergic denervation.
Authors: Gasparotto J, Ribeiro CT, Bortolin RC, Somensi N, Rabelo TK, Kunzler A, Souza NC, Pasquali MAB, Moreira JCF, Gelain DP.
Journal: Sci Rep. 2017 Aug 18;7(1):8795.
PMID: 28821831 (This article is OPEN ACCESS if you would like to read it)
In this study, the researchers wanted to see what would happen when they blocked RAGE in a model of Parkinson’s.
What is RAGE?
Receptor for advanced glycation endproducts (RAGE) is exactly what it says on the label – it is a molecule that sits on cell membranes and AGEs bind to it. On the surface of a cell, there are lots of receptors which act as switches for certain biological processes to be initiated. Receptors will wait for a protein to come along and activate them or alternatively block them.
The activators are called agonists, while the blockers are antagonists.

Agonist vs antagonist. Source: Psychonautwiki
RAGE is a receptor for AGEs. And it is by binding to RAGE that AGEs can activate signalling pathways within cells that are associated with both oxidative stress and inflammation, but also cell death.
Thus, the researchers wanted to see if they could rescue a model of Parkinson’s by blocking RAGE (not allowing AGEs to bind and activate the nasty signalling pathways).
They used a blocker of RAGE called ‘FPS-ZM1‘ (as far as I’m aware this drug is not being clinically developed – more on this below). They gave the animals this drug at the same time as a neurotoxin (6-OHDA) which kills dopamine neurons. The neurotoxin caused a 50% increase in RAGE activity in control (no RAGE inhibitor) animals, but the FPS-ZM1 drug reduced this effect by 46% in the treatment group.
The neurotoxin caused a significant loss of dopamine neurons in the control group which also resulted in motor behaviour impairments in the animals. FPS-ZM1 blocked the loss of dopamine neurons and also helped to rescue the motor problems resulting from the neurotoxin. The researchers concluded that their results demonstrate that RAGE is an essential component in the inflammatory process and dopamine cell loss associated with the delivery of the neurotoxin, and that selective inhibition of RAGE may offer interesting possibilities for therapeutic approaches.
And more recently, a study was published that suggests an even more specific involvement of RAGE in certain individuals with Parkinson’s:

Title: AGE-induced neuronal cell death is enhanced in G2019S LRRK2 mutation with increased RAGE expression.
Authors: Cho HJ, Xie C, Cai H.
Journal: Transl Neurodegener. 2018 Jan 23;7:1. doi: 10.1186/s40035-018-0106-z. eCollection 2018.
PMID: 29387348 (This article is OPEN ACCESS if you would like to read it)
Given that the Leucine-rich repeat kinase 2 (LRRK2) G2019S genetic mutation has been identified as one of the most prevalent genetic causes of late-onset Parkinson’s (Click here for more on this topic) and AGEs accumulate with age, the researchers behind this study wanted to investigate whether there were any interactions between AGEs and LRRK2.
What is LRRK2?
Also known as ‘Dardarin‘ (from the Basque word “dardara” which means “trembling”), LRRK2 is an enzyme that has many functions within a cell – from supporting efforts to move things around inside the cell to helping to keep the power on (involved with mitochondrial function).
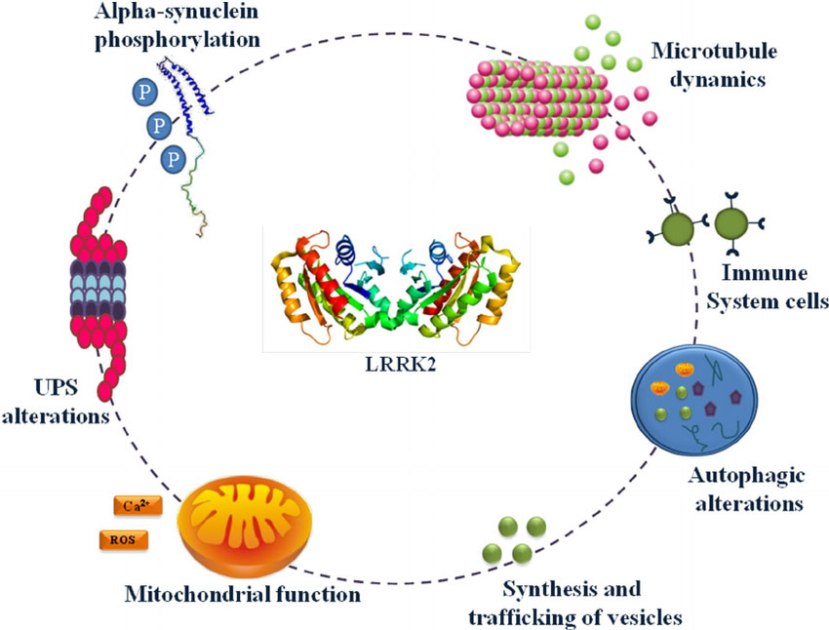
The many jobs of LRRK2. Source: Researchgate
The gene that provides the instruction for making the LRRK2 enzyme resides on the 12th chromosome, in an area of DNA referred to as ‘PARK8’ (one of the Parkinson’s-associated genetic regions). The LRRK2 gene is located within the PARK8 region, and it is made up of many different sections, each of which is involved with the different functions of the eventual protein.
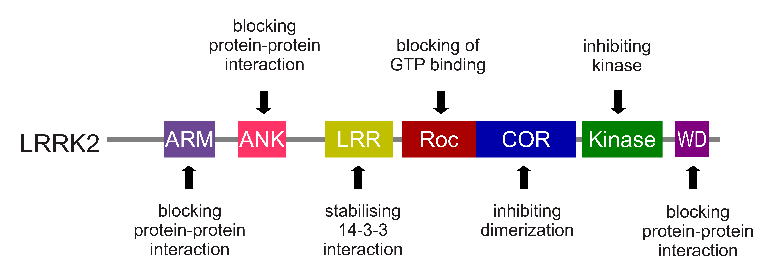
The regions and associated functions of the LRRK2 gene. Source: Intechopen
Genetic mutations within the LRRK2 gene are recognised as being some of the most common with regards to increasing ones risk of developing Parkinson’s (they are present in approximately 1-2% of all cases of Parkinson’s).
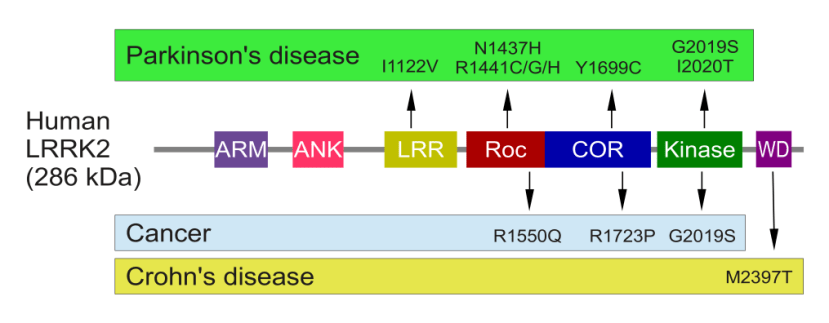
The structure of Lrrk2 and where various mutations lie. Source: Intech
As the image above suggests, mutations in the PARK8 gene are also associated with Crohn’s disease (Click here and here for more on this) – though that mutation is in a different location to those associated with Parkinson’s. And one particularly common Parkinson’s-associated LRRK2 mutation – called G2019S – is also associated with increased risk of certain types of cancer, especially for hormone-related cancer and breast cancer in women – Click here to read more about this. If you have a G2019S mutation, it is good to be aware of this association and have regular check ups.
The G2019S mutation (the name designates its location on the gene) is the most common LRRK2 mutation. In some populations of people it can be found in 40% of people with Parkinson’s (Click here to read more about this). But what is interesting about this mutation is that it gives rise to a LRRK2 enzyme that is hyperactive.
The structure of LRRK2 protein. Source: Wikipedia
As a protein, LRRK2 interacts with many different types of other proteins, and you can imagine that in a finely balanced environment like the cells that a mutant ‘hyperactive’ form of LRRK2 is going to cause problems. The consequences of this constantly active form of LRRK2 protein is believed to be the cause of cell death in LRRK2-associated Parkinson’s.
Getting back to this new study looking at AGEs and LRRK2, the researchers took cells with a LRRK2 G2019S genetic mutation and compared them to normal control cells. They found that the LRRK2 G2019S mutant neurons were more sensitive to AGE-induced cell death than the control cells, and this increased sensitivity in the LRRK2 G2019S mutant neurons could be reduced by blocking RAGE.
Importantly, the investigators found that the basal levels of RAGE proteins were higher in LRRK2 G2019S mutant cells. This means that there is more RAGE in the cells with the LRRK2 G2019S mutant regardless of whether any AGEs are present. And this increase in RAGE was also seen in the brains of mice that carry the LRRK2 G2019S mutant – at just 3 months of age (equivalent to early adulthood).
As a final experiment in their study, the researchers next looked at levels of RAGE in samples of postmortem brain from people with and with Parkinson’s.
And guess what they found?
Significantly increased levels of RAGE in the striatum from people who passed away with sporadic Parkinson’s (when compared to age matched control sample). The investigators concluded that “enhanced AGE-RAGE interaction contributes to LRRK2 G2019S mutation-mediated progressive neuronal loss in Parkinson’s”.
You mentioned blocking RAGE above. Are there any clinically available drugs for inhibiting RAGE?
No. Not yet.
The research described above suggests that inhibiting RAGE could be a beneficial approach to treating neurodegenerative conditions, like Parkinson’s. And there are biotech companies that are seeking to develop RAGE inhibitors.
The RAGE inhibitor that is furthest along in its development is called Azeliragon (also known as TTP488). A biotech company called vTv Therapeutics (formerly TransTech Pharma) is currently testing the drug in people with mild Alzheimer’s.
![]()
Azeliragon has been designed to bind to RAGE but not activate it, and thereby block AGEs from attaching to RAGE and activating the biological cascade that can lead to negative consequences. Azeliragon effectively turns RAGE off.
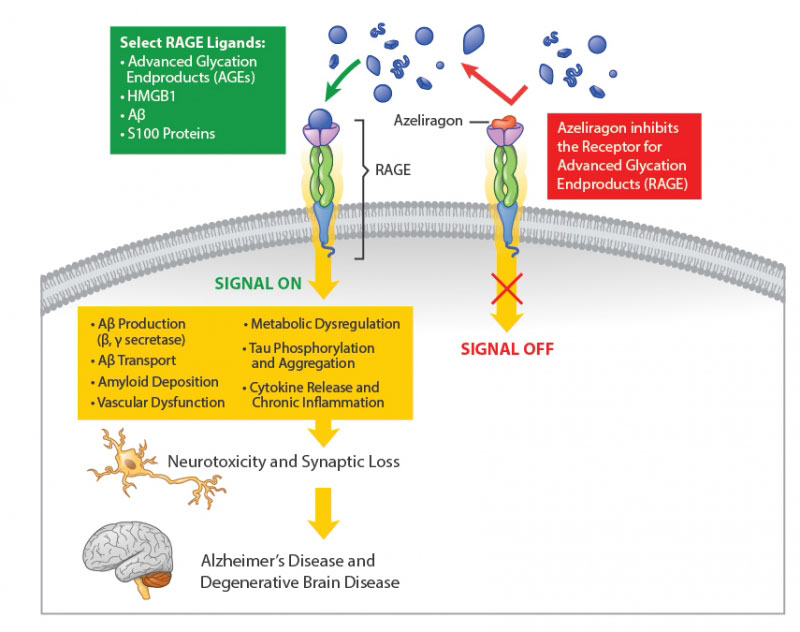
Source: vtvtherapeutics
vTv Therapeutics is currently conducting the Phase III STEADFAST study which is an efficacy and safety evaluation of Azeliragon in 800 people with mild Alzheimer’s. Participants in the study will receive either Azeliragon or placebo over a period of approximately 18 months (Click here to learn more about this clinical trial). The study should be completed by the end of this year and we will hopefully hear the results of the study in 2019, while awaiting the results of the follow on study which should be available in 2021 (Click here to learn more about that clinical trial).
The results of the first clinical trial of the drug have been published:

Title: Effect of TTP488 in patients with mild to moderate Alzheimer’s disease.
Authors: Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, Mjalli AM.
Journal: BMC Neurol. 2014 Jan 15;14:12. doi: 10.1186/1471-2377-14-12.
PMID: 24423155 (This study is OPEN ACCESS if you would like to read it)
In this first study, a Phase II, multi-center, double-blind, placebo-controlled clinical study (conducted at 40 study sites in the United States between January 2007 and December 2010), 399 patients were randomly assigned to one of two oral TTP488 (Azeliragon) doses (60 mg for 6 days followed by 20 mg/day; or 15 mg for 6 days followed by 5 mg/day) or a placebo treatment for 18 months. At the end of the study, the investigators found a significant treatment effects for participants taking 5 mg/day compared to those taking the placebo at month 18.

Source: Ncbi
As the graphs above suggest, the 5 mg/day TTP488 (Azeliragon) treated group demonstrated a slower decline based on the Mini Mental State Examination (MMSE) and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale test (ADAS-Cog) – both standardised tests for the evaluation of cognitive ability and memory.
What about the higher (20 mg/day) dose group?
Yeah…..um, about that.
Before anyone gets too excited about this drug, it should be noted that, at the 20 mg/day dose, some trouble was observed in this trial.
You never want to read the words “acute cognitive worsening” in a study report involving any drug being tested on dementia, but this is what occured in some of the participant receiving the 20 mg/day dose. And this study was originally conducted by our old friend Pfizer, which terminated their development programme for TTP488/Azeliragon after this particular result (and other results in type 2 diabetes ). All rights to the Azeliragon drug subsequently returned to TransTech Pharma (which rebranded itself as vTv Therapeutics). The dose window for Azeliragon is extremely narrow which makes one rather cautious about further investigations of this drug (and this has been noted in the research press – Click here for an example).
The key question is why would a high level of RAGE inhibition result in a worsening of cognitive ability? Does chronic RAGE inhibition cause cell death? Or is Azeliragon having some kind of off-target effect? Further research is obviously required.
Having said that, it will be interesting to see, however, how vTv Therapeutics gets on with the phase III trial of Azeliragon in mild dementia. If all goes well and some preclinical evidence suggesting a possible role for Azeliragon in Parkinson’s is reported in the near future, it would be interesting to investigate Azeliragon for use in Parkinson’s (perhaps as a supplemental treatment to neuroprotective agents in a multi-modal approach to treating Parkinson’s).
We’ll have to wait and see.
But while we wait, other groups are also exploring drugs that can inhibit RAGE. Recently a research group from the Diabetes Research Program in the New York University Langone Medical Center published research in which they identified 13 bona fide RAGE inhibitors from a screen of 58,000 small molecules (Click here to read that research report). They do not state what the compounds are, but we can be sure that they are now following up that research with the goal of taking RAGE inhibitors into clinical trials.
Can I naturally control my levels of AGE and RAGE?
Given their association with high levels of sugar, there are some obvious methods of reducing AGEs levels:
- Reducing sugar intake – This one is really simple. Eat fruit when you crave sugar.
- Exercising more – Muscles consume glucose, so the more muscle you have, the more glucose your body will want. As we age, we naturally lose muscle. Reduction in muscle mass can increase blood sugar levels, leading to increased AGEs. Regular weight training can help counteract this affect.
- Don’t throw shrimp on the barbie – Barbequing, searing, and broiling food can actually create AGEs in your food.
- Protect your skin – UV exposure increases the formation of AGEs in skin.
- Watch the alcohol intake – Researchers have found that alcohol enhances glycation stress.
So what does it all mean?
As we age, we build up AGEs – proteins that have haphazardly bound to sugar molecules which disrupts the ability of the protein to do it’s job properly. These AGEs can bind to a specific receptor (called RAGE) which activate biological pathways that can have negative impacts on our health.
Given that diabetes (a condition of high blood sugar levels) is associated with Parkinson’s, there has been a lot of research looking at AGEs & RAGE in the Parkinsonian brain and also models of Parkinson’s. Of particular interest is the presence of AGEs in Lewy bodies, and the interactions of AGEs with Parkinson’s-associated proteins, such as alpha synuclein and LRRK2.
It would be interesting to see what impact a RAGE inhibitor/blocker would have on Parkinson’s. By itself, a RAGE blocker may have a limited effect (as proteins like alpha synuclein continue to aggregate), but in combination with an alpha synuclein targeting drug/vaccine (Click here to read more about this), the effect may be more impactful. We will have to wait and see what happens with the currently available drugs being tested in the clinic before considering this dual-treatment approach.
Until then: Reduce your RAGE as you AGE. Sage advice for life.
The banner for today’s post was sourced from Dualshockers

Very nice that mentioned A.G.E.s
Main source AGE is Western diet with large amounts cooked meat.(grilled, broiled, fried, baked is way to make AGEs)
Best paper: “Advanced Glycation end Products in Foods and a Practical Guide to their reduction in the Diet, Uribarra, Vlassara, 2010.
Best book: Dr. Vlassara’s A.G.E.Less Diet (Dr Vlassara leading researcher last 30 years at Mt Sinai Hospital, NYC)
Link to my published paper: https://doi.org/10.18632/aging.101720
LikeLike
Hi Alan,
Thanks for your comment and for the information shared. Much appeciated.
Kind regards,
Simon
LikeLike