
|
The protein Alpha Synuclein has long been considered the bad-boy of Parkinson’s disease research. Possibly one of the main villains in the whole scheme of things. New research suggests that it may be interfering with a neuroprotective pathway, leaving the affected cell more vulnerable to stress/toxins. But that same research has highlighted a novel beneficial feature of an old class of drugs: MAO-B inhibitors. In today’s post we will outline the new research, discuss the results, and look at whether this new Trk warrants a re-think of MAO-B inhibitors. |

The great Harry Houdini. Source: Wikipedia
I’m not sure about you, but I enjoy a good magic trick.
That exhilarating moment when you are left wondering just one thing: How do they do that?
(Seriously, at 4:40 a baguette comes out of thin air – how did he do that?)
Widely believed to have been one of the greatest magicians of all time (Source), Harry Houdini is still to this day revered among those who practise the ‘dark arts’.
Born Erik Weisz in Budapest (in 1874), Houdini arrived in the US in 1878. Fascinated with magic, in 1894, he launched his career as a professional magician and drew attention for his daring feats of escape. He renamed himself “Harry Houdini” – the first name being derived from his childhood nickname, “Ehrie,” and the last name paying homage to the great French magician Jean Eugène Robert-Houdin. In 1899, Houdini’s act caught the eye of Martin Beck, an entertainment manager, and from there the rest is history. Constantly upping the ante, his feats became bolder and more death defying.
And the crowds loved him.
From stage, he moved on to film – ultimately starting his own production company, Houdini Picture Corporation. In addition, he was a passionate debunker of psychics and mediums, his training in magic helping him to expose frauds (which turned him against his former friend Sir Arthur Conan Doyle, who believed deeply in spiritualism).
This is all very interesting, but what does any of it have to do with Parkinson’s?
Magic runs deep in the Houdini family: Aron Houdini – the great, great, great nephew of Harry – is also a magician…

Aron Houdini. Source: Youtube
….but one who has been diagnosed with Parkinson’s (Click here to read his story).
The Houdinis, however, are not the topic of today’s post, merely the opening act.
The main event deals with a very different type of magic Trk.
Don’t you mean ‘trick’?
No, I meant ‘Trk’. It’s not a typo.
Tropomyosin receptor kinase (or Trk) is a receptor for a neurotrophic factor called Brain-derived neurotrophic factor (or BDNF). Neurotrophic factors (neurotrophic = Greek: neuron – nerve; trophikós – to feed) are chemicals produced in the brain that help to nourish the neurons and keep them alive. They achieve this fantastic function by binding to specific receptors, such as Trk.
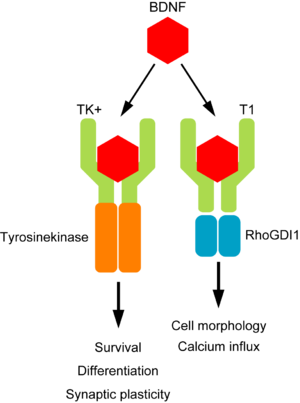
BDNF binding to receptors. Source: Wikipedia
Receptors are like the lock for a particular key.
Once the key is in the lock, specific processes are activated. In the case of BDNF (the key) and Trk (the lock), the activated processes are of a neuroprotective type. BDNF is very good at keeping neurons alive.
Thus, anything that affects BDNF signalling would obviously have a negative impact on a neuron’s ability to survive.
What things affects BDNF signalling?
Well, recently a protein that the Parkinson’s research community is very familiar with was found to disrupt BDNF signalling:

Title: TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death inParkinson’s disease.
Authors: Kang SS, Zhang Z, Liu X, Manfredsson FP, Benskey MJ, Cao X, Xu J, Sun YE, Ye K.
Journal: Proc Natl Acad Sci U S A. 2017 Sep 18. pii: 201713969.
PMID: 28923922
In this study, the researchers found the Parkinson’s associated protein Alpha Synuclein directly interacts with the TrkB receptor (there are three Trk receptors – A, B & C – and BDNF binds to TrkB).
But this interaction does not have many positive outcomes.
The investigators found that high levels of Alpha Synuclein reduces TrkB protein levels (as well as subsequent TrkB signaling), thereby inhibiting the neurotrophic effects of BDNF. And by inhibiting BDNF, Alpha Synuclein made the cells more vulnerable to neuronal cell death.
Interestingly, Alpha Synuclein was achieving this inhibition via several methods: Firstly, it was disrupting TrkB distribution on membranes. Second, it decreased its internalization once activated by BDNF. And it reduced its TrkB protein levels by causing the protein to be taken away by the waste disposal system.
Oh man! This is not good right? Not good at all.
Let’s hold our judgement until these results have been independently replicated, shall we? Many amazing results in research can not be repeated by other research groups, so until this result is validated it is unwise to start panicking.
What is very interesting in the findings of this study, however, is that the researcher identified the chemical that is stimulating the interaction between Alpha Synuclein and TrkB.
That chemical is called 3,4-Dihydroxyphenylacetaldehyde (or DOPAL).
What is DOPAL?
When your dopamine neurons release the chemical dopamine, the cell will quickly reabsorb any dopamine that is not used. That reabsorbed dopamine can then either be re-packaged for future release, OR it can be broken down for recycling/disposal.
The first step in that recycling process is conducted by an enzyme called Monoamine oxidase B (or simply MAO-B). It breaks dopamine down into DOPAL.

The recycling of old dopamine. Source: Google.sr
DOPAL is subsequently broken down into 3,4-dihydroxyphenylacetic acid (or DOPAC) by an enzyme called aldehyde dehydrogenase (ALDH). Interestingly, DOPAL levels have been found to be higher in Parkinsonian brains (Click here to read more about this), while ALDH levels have been found to be reduced in Parkinsonian brains (Click here for more on this).
DOPAL is generally considered a metabolite (or an intermediary substance formed during biological processes), but don’t for a second think that increased levels of DOPAL are a good thing or a non-event.
DOPAL is highly reactive (meaning it can cause oxidative stress) and it is thus toxic for cells. It is usually rapidly processed into DOPAC.
The fact that DOPAL is interacting with another bad guy in the Parkinson’s story is not really surprising though. And this interaction between DOPAL and alpha synuclein has recently been supported by the findings of other research groups:

Title: DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function.
Authors: Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M, Girotto S, Dalla Serra M, Perego C, Casella L, Bubacco L.
Journal: Sci Rep. 2017 Jan 13;7:40699.
PMID: 28084443 (This article is OPEN ACCESS if you would like to read it)
But if DOPAL is so bad, how do we stop it from forming?
Good question.
By inhibiting MAO-B, researchers are able to reduce the levels of DOPAL and subsequently increase the levels of dopamine floating around in neurons. If the dopamine isn’t being broken down and recycled, it can be re-used, right? And this idea has given rise to the use of a class of drugs called MAO-B inhibitors as a treatment option for Parkinson’s.
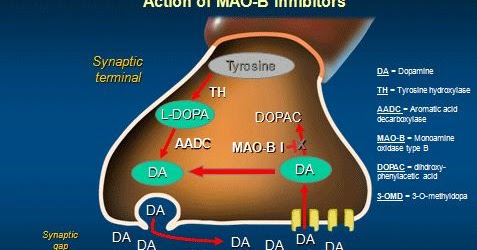
The cycle of dopamine production. Source: Geneticengineeringinfo
By treating people with MAO-B inhibitors, we can reduce the levels of DOPAL present in their dopamine neurons, which may in turn reduces the chances of a nasty interaction between Alpha Synuclein and DOPAL.
And this is exactly what the researchers behind the TrkB/BDNF study were thinking.
They treated both cells in culture that produce too much Alpha Synuclein and mice that produced too much Alpha Synuclein with the MAO-B inhibitor rasagiline. They found that the rasagline treatment disrupted the interaction between Alpha Synuclein and TrkB. This inhibition rescued BDNF’s neurotrophic activity. It also prevented dopaminergic neuronal death and restored motor functions in the mice.
The researcher concluded that their results provide “a model for the underlying molecular etiology of Alpha Synuclein-mediated neurotoxicity and dopaminergic neuronal loss in Parkinson’s disease”.
Does this result mean that rasagiline is neuroprotective?
So this is one of the most controversial topics in the field of Parkinson’s research. And it’s a big deal that completely divides investigators.
It is fair to say that the answer is not clear.
There have been two major clinical trials to try and determine whether rasagiline has any disease-modifying effects.
The first study was called the “TVP-1012 in Early Monotherapy for PD Outpatients” (or TEMPO) study:

Title: A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease.
Authors: Parkinson Study Group
Journal: Arch Neurol. 2004 Apr;61(4):561-6.
PMID: 15096406 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators conducted a randomised double-blind (meaning that no one knew who was getting what treatment), parallel-group, delayed-start clinical trial. The ‘parallel-group, delayed start’ design involved participants being randomly assigned to receive rasagiline (either 1 or 2 mg per day) for 1 year OR to receive placebo for 6 months followed by rasagiline (2 mg per day) for 6 months. These two groups being treated differently in parallel could then be compared at the end of the study to determine if the drug had any disease modifying effect – if there is an effect, the group that received rasagiline for the full 12 month should be better off than the group that had placebo for 6 months before being given rasagiline for 6 months (as a result of being treated with the drug for a longer period).
A total of 404 people with recently diagnosed Parkinson’s that was not being treated with dopamine-based therapy (for example L-dopa) were recruited at 32 different research sites across the United States and Canada. After the 12 month long study, the researchers found that the subjects receiving either dose of rasagiline (1 or 2 mg per day) for the whole year were better off by approximately 2 units on their Unified Parkinson’s Disease Rating Scale (UPDRS) scores when compared to the group that received the placebo plus rasagiline. This result suggests that there was an positive effect from taking the MAO-B inhibitor.
It was a minimal effect, but an effect nonetheless.
The second large clinical study that has looked at this issue was called the “Attenuation of Disease Progression with Azilect Given Once-daily” (or ADAGIO) study (Azilect being the commercial name for rasagiline):

Title: A double-blind, delayed-start trial of rasagiline in Parkinson’s disease.
Authors: Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F, Tolosa E; ADAGIO Study Investigators.
Journal: N Engl J Med. 2009 Sep 24;361(13):1268-78.
PMID: 19776408 (This article is OPEN ACCESS if you would like to read it)
This second study followed a similar design to the first study with a total of 1176 people with untreated Parkinson’s being randomly assigned to either receive rasagiline (at a dose of either 1 mg or 2 mg per day) for 72 weeks OR receive a placebo treatment for 36 weeks followed by rasagiline (at a dose of either 1 mg or 2 mg per day) for 36 weeks.
The results of the ADAGIO study were not as clear as the TEMPO trial. Only the group of subjects receiving 1 mg daily for the full study had significantly better UPDRS scores at the end of the study. The group receiving the higher dose of rasagiline (2 mg per day) for the entire study showed no different to the group that received the placebo treatment for the first 9 months of the study. The investigators concluded that rasagiline could possibly be having a disease-modifying effect in the 1 mg group.
My initial interpretation of this data is that there isn’t much evidence for a major disease modifying effect in the clinic for rasagiline in Parkinson’s disease.
This conclusion is confounded, however, by a 6.5 year follow up study of the TEMPO trial participants. That follow up study found that the participants that were in the ‘rasagiline for the whole trial’ group were 16% better off than the participants in the placebo-rasagiline treated group, even at 6 years after the trial was finished (Click here to read more about this follow up study).
You can see why this issue is so debated by member of the Parkinson’s research community. For an interesting OPEN ACCESS review of the MAO-B inhibitor clinical trials – Click here.
What does it all mean?
New research has identified a novel pathway via which cells could be made vulnerable by high levels of the Parkinson’s-associated protein Alpha Synuclein. By disrupting the neurotrophic effects of BDNF, Alpha Synuclein could be leaving the cells more vulnerable to injury or toxins. That same research has indicated that MAO-B inhibitors can reduce this damaging effect of Alpha Synuclein by reducing the protein that is stimulating the interaction between Alpha Synuclein and the BDNF receptor.
While the result is very interesting, the situation in the clinic does not fully support the findings. The minimal disease modifying effect observed in clinical trials of rasagiline thus far, are not extremely convincing. But those trials have largely tested MAO-B inhibitors as a mono-therapy for Parkinson’s, perhaps in concert with other treatments a neuroprotective effect could become more apparent. This is yet to be tested though.
Based on reading the results of the study we have reviewed today, there may be some temptation by readers to try a MAO-B inhibitor treatment. Before considering any actions, a physician familiar with their medication regime should be consulted. MAO-B inhibitors are known to conflict with many other classes of drugs, including most antidepressants. So caution is advised.
And one last thing: I have taken a bit of artistic licensing with this post. Trk is generally pronounced “Track” in the research community rather than “Trick”.
EDITORIAL NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from Youtube

I think I remember studies on selegiline too? How does this other MAO-B Inhibitor compare? thanks again
LikeLike
Hi Dkdc,
I hope all is well.
There are a few MAO-B inhibitors, but Selegiline and Rasagiline are the two main players. Both are relatively selective and irreversible MAO-B inhibitors, but there are some differences – mainly with regards to the bioproducts of their activities (for example, rasagiline produces aminoindane, while selegiline produces amphetamine derivatives). And it is this last detail that is key to further discussions – see, the amphetamine derivatives may actually inhibit cells ability to reabsorb dopamine, which obviously increases the levels of dopamine in service. This means that selegiline may have the dual effect of blocking the break down of dopamine (MAO-B inhibition) and also blocking the reabsorption of dopamine. This makes it difficult to determine if selegiline is really having a disease modifying effect, compared to Rasagiline which just blocks MAO-B (these sorts of comments are known to cause fights among researchers so I’ll stop here).
There have been several studies suggesting ‘disease modifying’ effects of selegiline (https://www.ncbi.nlm.nih.gov/pubmed/16540603; https://www.ncbi.nlm.nih.gov/pubmed/9749589; https://www.ncbi.nlm.nih.gov/pubmed/27911341), but whether these effects are disease modifying or simply due to that amphetamine derivative issue is hotly debated.
This year there has been a research report (https://www.ncbi.nlm.nih.gov/pubmed/27911341) about a study involving 1616 participants (784 on MAO-B inhibitors – 75% of those on Rasagiline and 17% on selegiline). That study found a “significant association between longer duration of MAO-B inhibitor exposure and less clinical decline”. I didn’t mention this in the post, however, as the study was a post-hoc analysis and not a randomized, blinded trial. But it may lend some credence to the idea that MAO-B inhibitors are doing more than simply inhibiting MAO-B.
I hope this helps,
Simon
LikeLike
Thanks
LikeLike
Thanks for deciphering stuff like this, Simon. I read somewhere that the variants of two snps were associated with symptomatic benefits with rasagiline and wonder if that’s why the overall effects seem so small. It would be interesting if they had genotype info for tempo and other study subjects.
LikeLike
Hi Beetheaven,
You’re welcome, and many thanks for the really interesting question. It has highlighted an aspect of this story that I wasn’t actually aware of, but could change one’s interpretation of the findings of these studies.
So, in May last year the researchers behind the ADAGIO trial (the study in which the 1mg dose had an effect, but not the 2mg dose) published a research report that looked at 204 genetic variants (single nucleotide polymorphisms – SNPs) in 692 available DNA samples from subjects that took part in their clinical trial. The analysis highlighted two variants in the dopamine D2 receptor gene (DRD2) which were significantly associated with a favourable peak response to rasagiline. Individuals with these variants had an earlier and larger improvement in Parkinson’s disease symptoms when treated with rasagiline compared those without the variants.
The research article is OPEN ACCESS if you would like to read it ( https://academic.oup.com/brain/article-lookup/doi/10.1093/brain/aww109). This is a really interesting finding, and it further justifies genotyping folks before recruiting them to particular drug trials (in order to help determine efficacy). It would be interesting to repeat these clinical trials with this new knowledge.
Great comment/question – many thanks!
Kind regards,
Simon
LikeLike
Hi Simon,
Thanks for yet another great post.
Do you think that L-Dopa treatment might actually cause Parkinson’s to progress faster? (L-Dopa -> more dopamine -> more DOPAL?)
LikeLike
Hi Knas,
Thanks for the interesting question – glad you liked the post.
It’s a popular idea – that L-dopa might actually be aiding the progress of the condition – but the empirical and clinical evidence hasn’t really supported it up to this point (at least not in any exaggerated way). For example, in general folks on dopamine agonists (which don’t involve any increase in DOPAL, etc) don’t really have a slower level of progression. In addition, postmortem pathological studies have not demonstrated any evidence of accelerated loss of dopamine neurons in folks who took L-dopa, when compared to dopamine agonists.
Having said that, the long-term side effects of L-dopa could be masking ongoing neuronal degeneration (particularly where doses are increasing). The ELLDOPA trial in the early 2000s hinted at this. The researchers behind that study gave recently diagnosed folks different doses of L-dopa for 40 weeks and then assessed them. If L-dopa is toxic, the people on the highest dose should be worse off. In the clinical assessments this was not the case, but in the brain imaging data there was evidence to suggest that levodopa might be accelerating the loss of dopamine nerve terminals. The researcher do note in that study that “although our imaging data showed greater decline among patients in the levodopa group, these data do not indicate a “levodopa-inflicted cell loss” but, as we said, may have several explanations” ( http://www.nejm.org/doi/full/10.1056/NEJMoa033447#t=article ). So even they are reluctant to say Yes or No L-dopa is (or isn’t) having an impact on disease progression.
Long story short: Currently there is little solid evidence to support the idea.
Kind regards,
Simon
LikeLike
Thank you Simon!
LikeLike
Thank you Simon! Excellent article.
LikeLike
Thank you for the info on Rasagiline. I was diagnosed with early onset PD at the age of 59 – 9 years ago. I’ll be 69 next month and have been taking (Azilect at the start) and then generic Rasagiline 1mg per day since February 2013. Within a few weeks of taking it in 2013 over 90% of my head tremors stopped! I can say my experience has been great with Rasagiline. I do not take any additional Rx, other than vitamins and supplements. My neurologist would tell you my PD has progressed very little, if at all in the last 9 years. I’m now retired and researching natural options to add to help myself. Considering adding Quercetin. Thank you to all the researchers working on PD!
To neurologists … I don’t understand why some neurologists put people immediately on Carbidopa Levadopa! I’ve watched friends who presented with similar early onset symptoms as me diagnosed and immediately given this drug that makes their symptoms far worse within a few months of taking it! More neurologists need to give Rasagiline and MAO-B inhibitors as monotherapy a chance for the benefit of their patients.
Thank you!
From a happier healthier PD patient due to Azilect/Rasagiline.
LikeLike
Hi Diane! I just came upon your post today. I was recently diagnosed with early stage Parkinson’s (tremor dominant). I’m 54. I have left side tremors in my hand/arm and leg; mild bradykinesia of my left hand and some mild left arm/shoulder/hand rigidity. My mobility and balance are excellent and I exercise daily with a variety of exercises (Rock Steady Boxing, fast walking on a treadmill, Smoovey vibrating rings, weight lifting, and exercise/stretches with physical therapy resistance bands).
My PD/movement disorder specialist does not think I need any medication at this time, but he suggested that I consider taking Azilect (alone) since it MIGHT have some disease modifying properties. I am uncertain about taking it. Some of the possible side effects sound horrible and scary. And generally, I like to avoid medications! I have been looking for posts/info from people who have had positive experiences with Rasagiline. So I read your post with great interest! Are you still doing well only on the Rasagiline? Do you do other things (like exercise) to help stall the progression? Thanks for your time! Tracy Tully
LikeLike
Good evening Tracy,
Yes! I’m still doing great on Rasagiline only. I would definitely give it a try if I were you. I do try to exercise, walk and work a lot in my yard with veggie and flower garden. I also take various supplements. One I’ve added daily, since having Covid in Jan. 2021, is Quercetin, which I take with vitamin C as I understand it helps the bioavailability of Quercetin. I understand Quercetin is a natural antihistamine in addition to being a great antioxidant. With Rasagiline you can’t have most decongestants, and the Quercetin, I think, immediately takes away any stuffy or runny nose … haven’t had one for more than a day in two years. And hopefully won’t for a while longer! So do look into Quercetin with Rasagiline for Parkinson’s.
There are several food side effects I find with Rasagiline … more now than I had years ago. High tyramine foods can interact with Rasagiline and raise your heart rate slightly. This bothers me mostly in the evening if I have high tyramine foods. So I’ve given up pepperoni on pizza, and anything with soy sauce, and try not to have chocolate in the evening or my heart rate stays up near 80-100 until midnight-2am.
Which makes it harder to fall asleep. I really just need to eat healthier and I’d be great! I haven’t had any other side effects that I’m aware of. Praying you will do as well as I’m doing after 10yrs on Rasagiline! I truely believe it’s delaying the progression of my Parkinson’s. 🙏🤗❤️💐🎶
LikeLike