
|
# # # # New approaches for potentially slowing the progression of Parkinson’s are being announced on a regular basis. Some of them can not be independently replicated (such is the nature of science), while others open up whole new areas of research. Recently scientists have reported that inhibiting certain aspects of the kynurenine pathway – which plays a critical role in generating energy in cells – can have neuroprotective results in models of Parkinson’s. Many of the results have been independently replicated and the findings are now resulting in a new class of drug heading for clinical testing. In today’s post, we will delve into what the kynurenine pathway is, explore how it relates to Parkinson’s, and discuss some of the approaches soon heading for the clinic. # # # # |
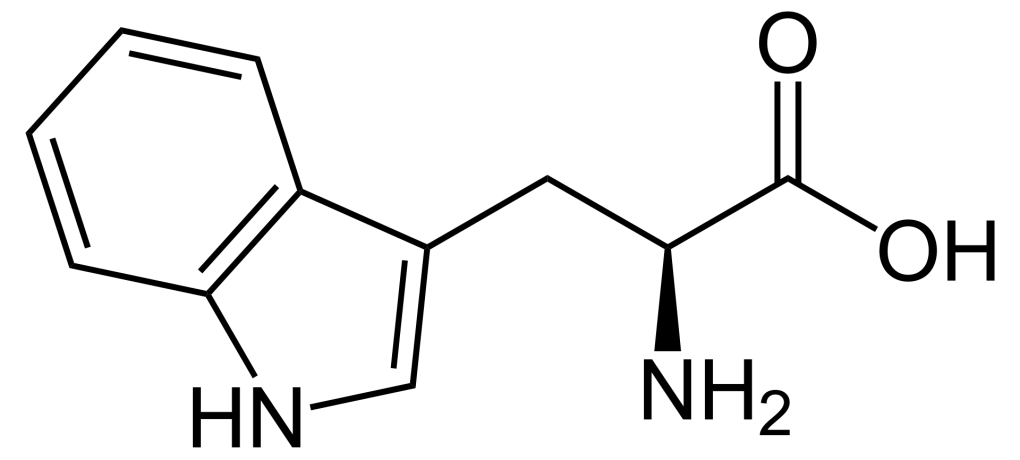 Structure of tryptophan. Source: Wikipedia
Structure of tryptophan. Source: Wikipedia
Tryptophan is one of eight essential amino acids.
Amino acids are the fundamental building blocks of proteins in biology, but the “essential” label in this case does not refer to its necessity (although it is necessary), but rather the fact that it cannot be made by our bodies. As a result, all essential amino acids must come from the food we consume.
Tryptophan has many functions within the body:
- it is a precursor to the neurotransmitter serotonin (which influences your mood, cognition, and behaviour)
- it is a precursor of the hormone melatonin (which governs your sleep-wake cycle)
- it is a precursor of vitamin B3 (naicin)
 Source: Wikimedia
Source: Wikimedia
More importantly, however, tryptophan is also involved in kynurenine synthesis.
What is kynurenine synthesis?
The kynurenine pathway plays a critical role in generating energy in cells – in the form of nicotinamide adenine dinucleotide (NAD+ – click here to read a previous SoPD post about NAD). Thus, the kynurenine pathway is a really important biological process.
To demonstrate how important this pathway is: In the human brain, approximately 95% of tryptophan is involved in the kynurenine pathway, while the remaining tryptophan is used for the synthesis of serotonin and melatonin.
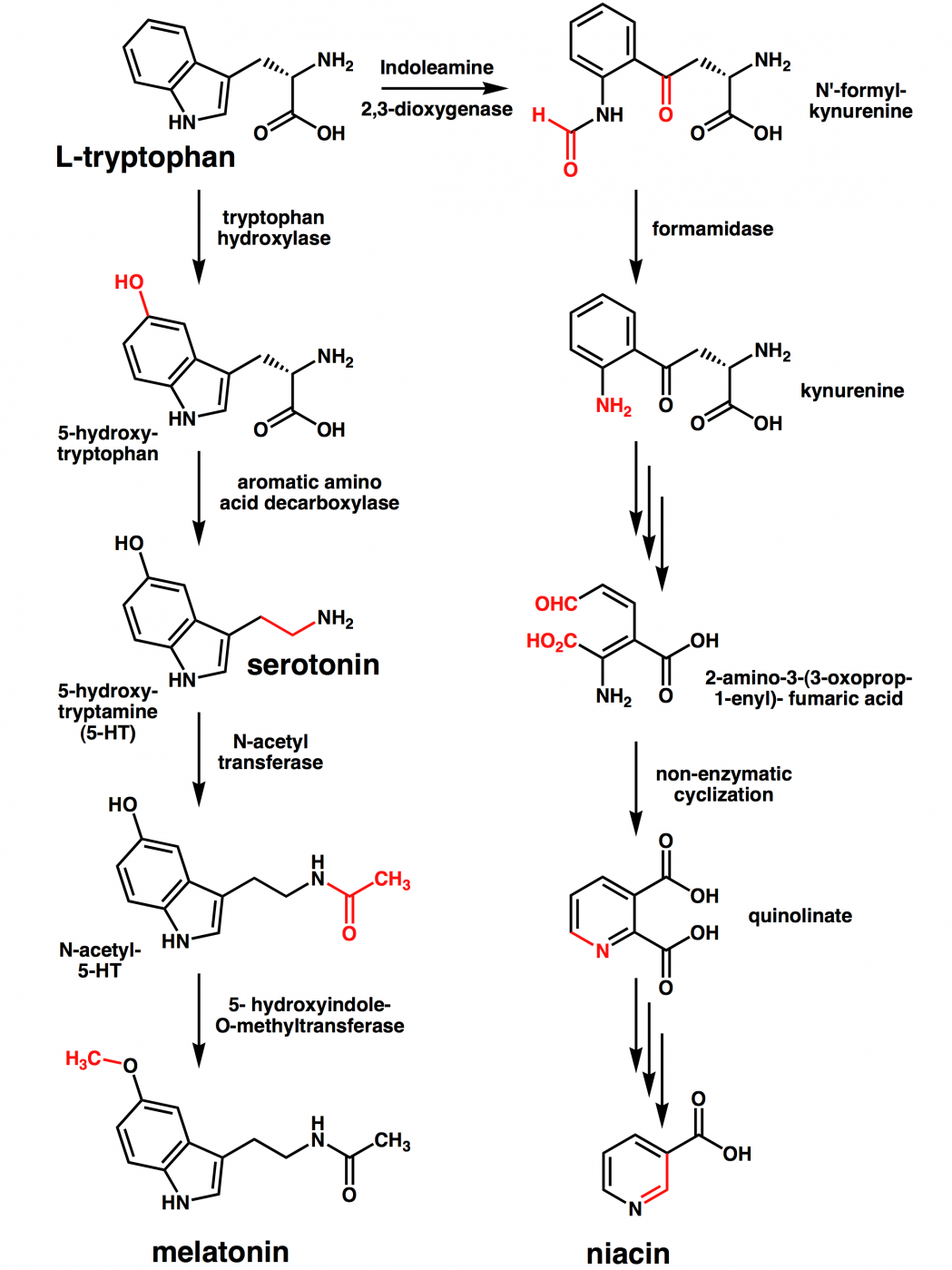
Like many biological pathways, the kynurenine pathway involves a cascade of steps and reactions, which results in many intermediaries (also called metabolites). Many of these metabolites are themselves neuroactive, including 3-hydroxykynurenine (3-HK), quinolinic acid (QUIN) and kynurenic acid (KYNA).
 The kynurenine pathway. Source: Enamine
The kynurenine pathway. Source: Enamine
It is intriguing that some of these metabolites are well-characterized endogenous neurotoxin that can cause excitotoxicity or oxidative stress (such as QUIN and 3-HK), while others have demonstrated neuroprotective properties (for example KYNA).
It is also interesting to note that in many neurodegenerative conditions, there are apparent shifts towards an increased synthesis of the neurotoxic metabolites QUIN and 3-HK compared to KYNA, which may contribute to the progression of these conditions. For example, 3-HK and QUIN levels have been reported to be increased and KYNA levels have been shown to be reduced in the brains of people diagnosed with Huntington’s disease (Click here and here to read more about this).
What about Parkinson’s? What happens to QUIN, 3-HK and KYNA in the PD brain?
Similar changes have also been reported in PD. For example, this report demonstrated a shift in the brains of people with Parkinson’s:
 Title: Kynurenine pathway abnormalities in Parkinson’s disease.
Title: Kynurenine pathway abnormalities in Parkinson’s disease.
Authors: Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S.
Journal: Neurology. 1992 Sep;42(9):1702-6.
PMID: 1513457
In this study, the researchers measured metabolites of tryptophan in various regions of the brain. They analysed the postmortem brains of people with and without Parkinson’s, and they found that levels of the potentially neurotoxic 3-hydroxykynurenine (3-HK) were increased in the putamen and substantia nigra of the Parkinson’s brain – both of these areas are severely affected by PD.
The investigators also observed a decrease in levels of kynurenic acid (KYNA) – which as mentioned above has neuroprotective potential. The authors noted in their report that “since the KYNA pathway leads to formation of nicotinamide-adenine dinucleotide (NADH), the present results may be a further indication of a defect in” mitochondrial function in PD (mitochondria being the small power stations inside of cells).
A second report – in which cerebrospinal fluid was collected from postmortem brains of people with and without Parkinson’s – reported a one-third increase in levels of 3-HK (Click here to read more about this).
And a more recent report has explored the kynurenine pathway in the context of individuals with Parkinson’s and L-DOPA-induced dyskinesia.
This is that report:

Authors: Havelund JF, Andersen AD, Binzer M, Blaabjerg M, Heegaard NHH, Stenager E, Faergeman NJ, Gramsbergen JB.
Journal: J Neurochem. 2017 Sep;142(5):756-766.
PMID: 28628213 (This report is OPEN ACCESS if you would like to read it)
The researchers collected blood and cerebrospinal fluid samples from 14 unaffected individuals (controls), 8 people with Parkinson’s who were not receiving no L‐DOPA, 8 people with Parkinson’s being treated with L‐DOPA but without dyskinesia, and 10 people with Parkinson’s and L‐DOPA‐induced dyskinesia.
The investigators found large increases in levels of 3-HK in both the blood (plasma) and cerebrospinal fluid (CSF) samples in the people with Parkinson’s and L‐DOPA‐induced dyskinesia.
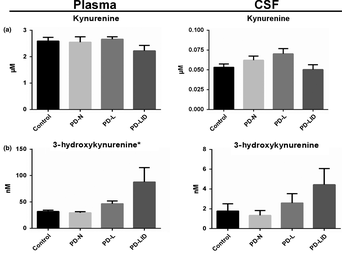 Source: Wiley
Source: Wiley
This result left the researchers asking if 3-HK could “serve as a biomarker for L‐DOPA‐induced dyskinesia” , and they proposed larger, longitudinal studies to further evaluate this possibility.
|
# RECAP #1: Tryptophan is an essential amino acid that is involved in the kynurenine pathway. Kynurenine plays a critical role in generating energy in cells. Intermediates (also called metabolites) in the biological processes of the kynurenine pathway have been found to have positive and negative properties. The balance of these metabolites is shifted in the brain in some cases of Parkinson’s. # |
Can this balance of the metabolites be corrected?
So this is a question that a number of researchers have been asking.
And a report was published in 2012 exploring this idea:
 Title: Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation.
Title: Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation.
Authors: van der Goot AT, Zhu W, Vázquez-Manrique RP, Seinstra RI, Dettmer K, Michels H, Farina F, Krijnen J, Melki R, Buijsman RC, Ruiz Silva M, Thijssen KL, Kema IP, Neri C, Oefner PJ, Nollen EA.
Journal: Proc Natl Acad Sci U S A. 2012 Sep 11;109(37):14912-7.
PMID: 22927396 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers identified an enzyme called tryptophan dioxygenase (TDO) as a metabolic regulator of age-related alpha synuclein toxicity in a Caenorhabditis elegans model.
Wait. What does any of that mean?
Ok, let’s start with tryptophan 2,3-dioxygenase.
This is one of three enzyme that can degrade tryptophan – the three are:
- tryptophan dioxygenase (TDO)
- indoleamine-2,3-dioxygenase (IDO)
- indoleamine-pyrrole 2,3-dioxygenase-like 1 (INDOL 1).
These enzymes allow for tryptophan to be used in the production of various metabolites in the kynurenine pathway.
And in this report, the investigators found that the tryptophan enzyme “TDO” is a regulator of age-related alpha synuclein toxicity.
We have discussed the Parkinson’s-associated protein alpha synuclein a lot on this website (Click here to read a previous SoPD post about this). Briefly, alpha synuclein is one of the most common proteins in the brain, but for some reason in Parkinson’s it starts to clump together and form “aggregates”. And these clusters of alpha synuclein protein are believed to be involved in the cell death associated with PD.
Ok, but what is a Caenorhabditis elegans model?
Caenorhabditis elegans (or simply C. elegans) are transparent nematode – also known as roundworms. They are about 1 mm in length, and they have an extremely well characterised nervous systems.
Absolutely useless pub quiz facts: C. elegans have 302 neurons and 56 glial cells in total, which communicate through approximately 6400 chemical synapses, 900 gap junctions, and 1500 neuromuscular junctions (like I said, they are very well characterised!).
 Caenorhabditis elegans – cute huh? Source: Nematode
Caenorhabditis elegans – cute huh? Source: Nematode
Given their thoroughly mapped out nervous systems, C. elegans provide a useful tool for studying biology. They are easy to grow/maintain, they have an overall life span of 2-3 weeks, and researchers have developed a wide range of tools that allow for genetic manipulation to address specific questions.
In the current study, the researchers used C. elegans that produced large amounts of alpha synuclein in their neurons, which ultimately lead to cell death as the creatures age. To determine which genetic factors influence the toxicity of this model, the investigators started manipulating 80 regions of DNA (called genes) and they found 10 genes that increased the toxicity. But of interest to us is that they also identified 3 genes that decreased alpha synuclein toxicity.
And one of those was TDO.
Curiously, reducing levels of TDO in the C. elegans also suppressed the toxicity of other aggregation-prone proteins, including Alzheimer’s-associated beta-amyloid. This finding suggested to the researchers that TDO may function as a general regulator of protein homeostasis. Further analysis indicated that the ability of TDO to suppress alpha synuclein toxicity was independent of downstream metabolites in the kynurenine pathway.
Next the researchers wanted to test whether the accumulation of tryptophan was involved in regulating proteotoxicity, rather than TDO itself. By supplementing the food of the C. elegans with increasing amounts of tryptophan, the investigators observed a dose-dependent suppression of the alpha synuclein toxicity. This result suggested to them that the inhibition of TDO regulates proteotoxicity by increasing tryptophan levels.
The researchers concluded their study by suggesting that “tryptophan metabolism may offer avenues to reducing proteotoxicity in aging and age-related diseases”.
Interesting. Has anyone else reported similar results?
Yes. The study above was conducted by researchers from the Netherlands, France and Germany, but another independent group of scientists from the University of Leicester in the UK (along with US collaborators) reported similar results in 2016.
This is their report:
 Title: Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites.
Title: Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites.
Authors: Breda C, Sathyasaikumar KV, Sograte Idrissi S, Notarangelo FM, Estranero JG, Moore GG, Green EW, Kyriacou CP, Schwarcz R, Giorgini F.
Journal: Proc Natl Acad Sci U S A. 2016 May 10;113(19):5435-40.
PMID: 27114543 (This report is OPEN ACCESS if you would like to read it)
In this study, the investigators used genetically engineered flies to show that inhibition of two kynurenine pathway enzymes (TDO and kynurenine-3-monooxygenase) reduced neurodegeneration in models of Parkinson’s, Huntington’s, and Alzheimer’s.
What is kynurenine-3-monooxygenase?
Kynurenine-3-monooxygenase (or simply KMO) is also an enzyme in the kynurenine pathway.
It is involved with converting kynurenine to 3-hydroxykynurenine (3-HK – which is one of the metabolites with more negative properties that we discussed above):
 Source: Semanticscholar
Source: Semanticscholar
So TDO converts tryptophan into kynurenine and KMO converts kynurenine into 3-HK?
Essentially yes.
As a result, KMO inhibition functions via a more specific mechanism to the broader TDO inhibition.
And by inhibiting KMO or TDO in fly models of Parkinson’s, Huntington’s and Alzheimer’s, the researchers were able to reduce the cell death and behavioural features associated with these models – providing supportive data for the previous report suggesting TDO inhibition is neuroprotective and highlighting a second kynurenine pathway-related mechanism that has neuroprotective potential.
But has all of this research only been conducted in worms and flies?
No. Very recently (January 2021) the researchers in the Netherlands have followed up their previous research (discussed above) by testing a brain penetrant TDO inhibitor (called NTRC 3531‐0) in mouse models of Parkinson’s, and the results are (sorry Chris) interesting:
 Title: Pharmacological validation of TDO as a target for Parkinson’s disease.
Title: Pharmacological validation of TDO as a target for Parkinson’s disease.
Authors: Perez-Pardo P, Grobben Y, Willemsen-Seegers N, Hartog M, Tutone M, Muller M, Adolfs Y, Pasterkamp RJ, Vu-Pham D, van Doornmalen AM, van Cauter F, de Wit J, Gerard Sterrenburg J, Uitdehaag JCM, de Man J, Buijsman RC, Zaman GJR, Kraneveld AD.
Journal: FEBS J. 2021 Jan 20. Online ahead of print.
PMID: 33471408 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers wanted to evaluate TDO inhibition as a potential therapeutic strategy for Parkinson’s. To do this, they developed their brain penetrant TDO inhibitor NTRC 3531‐0 and compared it to another TDO inhibitor (called LM10 – Source) in a neurotoxin (rotenone) model of Parkinson’s.
The investigators reported that both TDO inhibitors reduced the severity of motor issues in the mice. TDO inhibitor treatment was also associated with decreased loss of dopamine neurons and reduced microglia activation in the substantia nigra region of the brain. In addition to this, the TDO inhibitors reduced accumulation of alpha synuclein in the enteric nerves, which surround the intestinal tract. And these results are made more encouraging by the fact that treatment with the inhibitors was initiated a week after the induction of the PD model.
The researchers concluded their report by writing that their “study shows for the first time in a relevant animal model that TDO inhibition with small molecule drugs may be beneficial for treatment of PD symptoms”.
Is anyone developing TDO inhibitors for clinical testing?
As far as I’m aware there are no TDO inhibitors being clinically tested for Parkinson’s at present (TDO inhibition is also being explored for oncology – click here for a review of this topic).
That said, the Dutch researchers who conducted the study mentioned above work at Netherlands Translational Research Center.
 Source: NTRC
Source: NTRC
And the researchers at NTCR appear to be keen “to optimize this newly developed TDO inhibitor” over the next few years – perhaps in preparation for clinical testing (Source). So maybe in the not too distant future we will see movement towards the clinic for this new class of drug.
|
# # RECAP #2: Inhibition of enzymes involved in the kynurenine pathway (such as tryptophan dioxygenase, or TDO) regulates proteotoxicity, which has a neuroprotective effect in models of neurodegeneration (including Parkinson’s). Researchers are now optimising orally available inhibitors of TDO in the hope of taking them forward into clinical testing. # # |
Ok, so that is TDO inhibition, but what about the KMO inhibition you mentioned above? Are people exploring that further?
Yes.
Back in 2005, a large genomic screen in yeast cells identified kynurenine 3-monooxygenase (or KMO) as a potential therapeutic target for the neurodegenerative condition of Huntington’s disease (Click here to read that initial report).
 Yeast cells. Source: NewEuropeans
Yeast cells. Source: NewEuropeans
This finding was supported was supported by reports that prolonged administration of relatively high doses of kynurenine (which leads to increased brain KYNA) rescues models of neurodegeneration (Click here and here to read examples of this).
And the result was further supported by this report in 2011 describing the characterisation of a KMO inhibitor:
 Title: Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration.
Title: Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration.
Authors: Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, Lee J, Truong J, Andrews-Zwilling Y, Hsieh EW, Louie JY, Wu T, Scearce-Levie K, Patrick C, Adame A, Giorgini F, Moussaoui S, Laue G, Rassoulpour A, Flik G, Huang Y, Muchowski JM, Masliah E, Schwarcz R, Muchowski PJ.
Journal: Cell. 2011 Jun 10;145(6):863-74.
PMID: 21640374 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers wanted to evaluate a novel KMO inhibitor called JM6 in a mouse model of Alzheimer’s. They found that administering JM6 to genetically engineered mice (which accumulate clusters of Alzheimer’s-associated beta amyloid in the brain from an early age) prevented memory deficits, anxiety-related behavior, and loss of synaptic connections in the brain.
They also reported that JM6 prevented the loss of synaptic connections in the brains of a mouse model of Huntington’s (R6/2 mice), and extended their life span (see image below):
 Source: PMC
Source: PMC
But then the researchers noticed something rather unexpected: JM6 does not get across the blood-brain barrier.
What is the blood-brain-barrier?
The blood-brain barrier is a protective membrane that surrounds the brain, separating circulating blood from the cerebrospinal fluid that the brain sits in. It is made up of endothelial cells that are connected by tight junctions. These cells limit the ability of many molecules (particularly large ones) to access the brain, making it a ‘barrier’ for many medications.
 The blood-brain-barrier. Source: Bioninja
The blood-brain-barrier. Source: Bioninja
Since JM6 was not entering the brain very well, this indicated that peripheral inhibition of KMO – that is inhibiting KMO outside of the brain – alone may be sufficient to confer neuroprotection. The researchers suggested that this may be an appealing approach going forward to the clinic, as it allows for modest and sustained elevations of KYNA levels in brain without having any unexpected negative effects.
But this finding has not stopped researchers from exploring brain penetrant KMO inhibitors though. An example of that research was published in 2019:
 Title: A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites.
Title: A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites.
Authors: Zhang S, Sakuma M, Deora GS, Levy CW, Klausing A, Breda C, Read KD, Edlin CD, Ross BP, Wright Muelas M, Day PJ, O’Hagan S, Kell DB, Schwarcz R, Leys D, Heyes DJ, Giorgini F, Scrutton NS.
Journal: Commun Biol. 2019 Jul 24;2:271.
PMID: 31372510 (This report is OPEN ACCESS if you would like to read it)
In this study, the investigators screened a library of 1000+ molecules for their suitablity as potential KMO inhibitors. That experiment resulted in 19 new KMO inhibitors (IC50 ≤ 200 µM). Further characterisation of these molecules allowed the researchers to zero in on one in particular (conveniently labelled an molecule #1), which was neuroprotective in a fly model of Huntington’s disease.
But then the investigators realised that molecule #1 had minimal brain penetrance in mice – it was not crossing the blood-brain barrier.
To circumvent this issue, the scientists turned their attention to the “prodrug” of molecule #1 (conveniently called molecule #1b), which does crosses the blood–brain barrier.
What is a produg?
A prodrug is a molecule that, after being administrated into the body, is converted (metabolised) via biological processes into the pharmacologically active drug of interest.
The researchers found that molecule #1b could cross the blood-brain barrier and then be converted into molecule #1, releasing it into the brain and allowing it to lower levels of 3-HK, the kynurenine pathway metabolite with toxic properties linked to neurodegeneration.
 Source: PMC
Source: PMC
The researchers concluded their study by stating that “Prodrug 1b will advance development of targeted therapies against multiple neurodegenerative and neuroinflammatory diseases in which kynurenine pathway likely plays a role, including Huntington’s, Alzheimer’s, and Parkinson’s disease”.
So what does it all mean?
A lot of the research discussed on this website is of varying degrees of proximity to potential clinical utility. Some posts are based on rather futuristic/”blue sky” research that may take at least a decade to translate into the clinic (or possibly never), while other posts are discussing re-purposable molecules which may be clinically available in a few years. The point is: there is a broad, rich spectrum of research involving many different approaches being evaluated for Parkinson’s, all at different stages of development.
The research discussed in today’s post is somewhere in the middle of that spectrum.
Researchers have determined that manipulating components of the kynurenine pathway results in neuroprotection, and they have identified novel molecules that they are now developing and optimising for clinical testing in the not too distant future. There are still lots of questions that need to be addressed regarding these classes of therapeutics before the leap to the clinic is made, but it is encouraging to learn of yet more approaches that are being explored to tackle Parkinson’s.
We’ll be on the look out for TDO and KMO inhibitors here at SoPD HQ.
Watch this space.
 All of the material on this website is licensed under a
All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
The banner for today’s post was sourced from Safebytes

{kind=link}
OK Simon, I’ll grant you this one!
Chris.
LikeLike
So, should we eat more turkey?
LikeLike
and another gene in the Kynurenine pathway is also neuroprotective https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6176922/
LikeLike
How do we know that the decreased levels of KYNA found in postmortem PD brains are not due to an increased need (and consumption) of it for energy production (e.g., to a need to defend the cell against microglial autoimmune attacks, or to garbage-collect alpha-synuclein, etc.), rather than to underproduction of it due to defects in the kynurenine pathway?
LikeLike