
|
Every textbook written about the condition will tell you that the classical pathological characteristic of Parkinson’s is the loss of dopamine neurons in the midbrain region of the brain. It is the distinguishing feature that pathologists look for in order to provide a postmortem diagnosis of the condition. But what is meant by the words ‘loss of dopamine neurons’? Do the cells actually die? Recently researchers from Korea have published new data exploring this question. Interestingly, they found evidence of ‘dormant’ dopamine neurons in postmortem sections of brains from people with Parkinson’s – even those with severe forms of the condition. In today’s post, we will discuss what a dopamine neuron is, what this new research found, and what it could mean for our understanding of Parkinson’s.
|
![]()
Source: Bettys
2019 represented the centenary year for an important discovery in Parkinson’s research.
In 1919, the Uzbek neuropathologist Konstantin Tretiakoff (1892-1958) reported his findings regarding an examination of 54 human brains.
 Konstantin Tretiakoff. Source: Wikipedia
Konstantin Tretiakoff. Source: Wikipedia
Six of the postmortem brains had belonged to individuals who had suffered from Parkinson’s and three others had been diagnosed with postencephalitic Parkinsonism. In these brains he noticed something rather striking.
What did he find?
In all of the Parkinson’s and postencephalitic Parkinsonism brains, Tretiakoff had reported a marked loss of pigmented neurons in a region called the substantia nigra (he also noted “Lewy bodies” – which Frederic Lewy had discovered just 9 years earlier – in the surviving neurons).
Source: Kingscollection
This was a major moment in Parkinson’s research and the finding has come to be a defining pathological feature of the Parkinsonian brain.
When a pathologist looks at a postmortem brain from a person who was diagnosed with Parkinson’s, they will immediately look at the substantia nigra to assess the loss of dopamine neurons. In a healthy brain, the dopamine neurons of the substantia nigra are visible with the naked eye, because they produce a dark pigment called neuromelanin (in the image below, you can see a bird’s eye view of the midbrain where the substantia nigra is located. On both sides of the section of brain on the left you can see the dark region that makes up the substantia nigra – labelled).
 The dark pigmented dopamine neurons in the substantia nigra are reduced in the Parkinsonian brain (right). Source: Memorangapp
The dark pigmented dopamine neurons in the substantia nigra are reduced in the Parkinsonian brain (right). Source: Memorangapp
In the Parkinsonian brain, however, these dopamine neurons are lost, which results in a reduction of this dark pigmentation (see brain on the right in the image above).
Interesting, but um, one question: what exactly is a dopamine neuron?
Good point.
Dopamine is a protein is the brain that plays a role in many basic functions of the brain, such as motor co-ordination, reward, and memory.
It works as a signalling molecule – a way for brain cells to communicate with each other. Dopamine is released from neuron, and it binds to target neurons, initiating biological process within those cells.
In this manner, it is called a neurotransmitter.
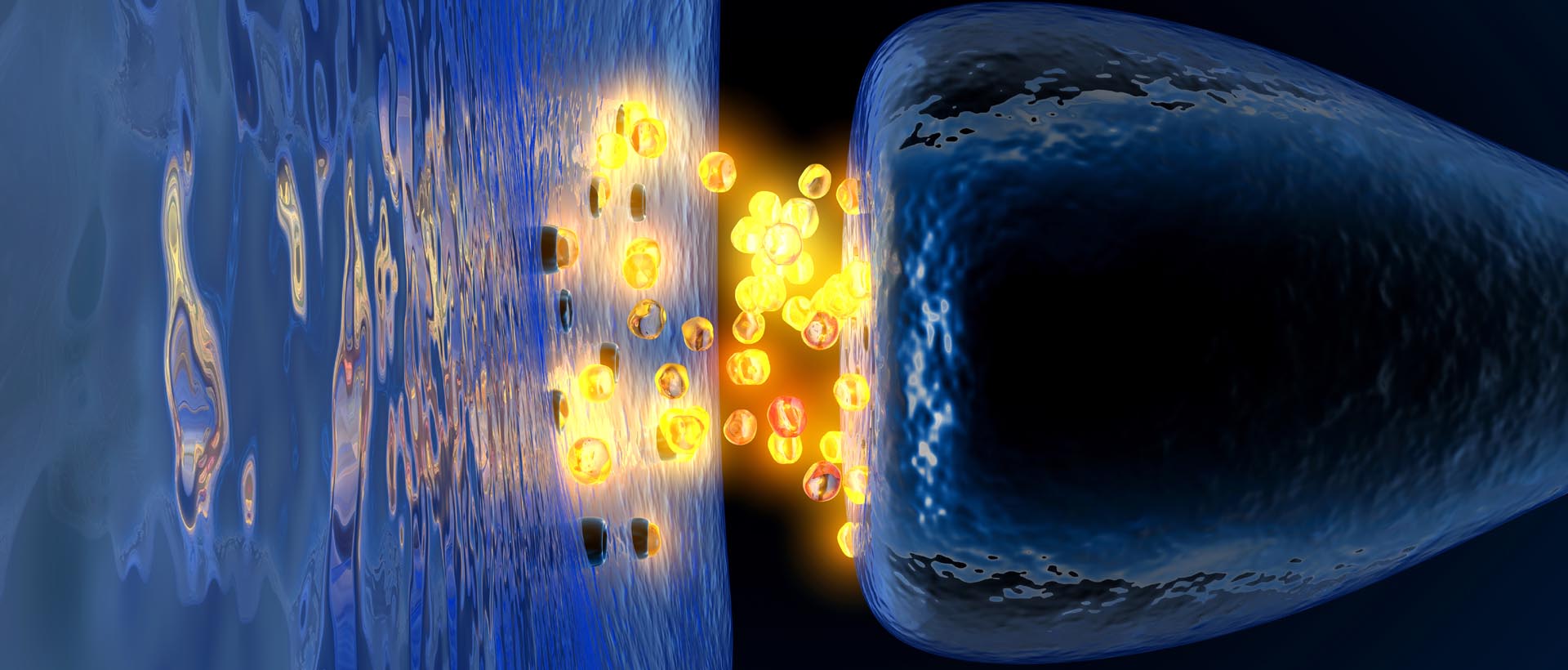 Dopamine being released by one cell and binding to another. Source: Truelibido
Dopamine being released by one cell and binding to another. Source: Truelibido
The dopamine neurons in the substantia nigra generate dopamine, but they release most of it in different areas of the brain. The primary regions of that release are areas of the brain called the putamen and the caudate nucleus. The dopamine neurons of the substantia nigra have long projections (or axons) that extend a long way across the brain to the putamen and caudate nucleus, so that dopamine can be released there.

The projections of the substantia nigra dopamine neurons. Source: MyBrainNotes
In Parkinson’s, these ‘axon’ extensions that project to the putamen and caudate nucleus gradually disappear as the dopamine neurons of the substantia nigra are lost. This results in a reduction of dopamine being released in these areas, which leaves the movement appears of the brain gradually becoming more inhibited. And this loss of dopamine and increase in inhibition shows itself clinically by the slowness of and problems initiating movement.
How do the dopamine neurons die?
The simple truth is: we don’t know.
But a more important question is: Do the dopamine neurons affected by Parkinson’s actually die?
|
RECAP #1: Dopamine is a protein that the brain produces and it plays an important role in passing signals from one cell to another. It is particularly important in modulating movement related signals in the brain. Parkinson’s is a neurodegenerative condition characterised by the loss of dopamine neurons. As the cells are lost, the movement regions of the brain become more inhibited, resulting in issues initiating movement. But do the all of the dopamine neurons actually die?
|
Do the dopamine neurons actually die?
To be clear: yes, in Parkinson’s there is neurodegeneration of the dopamine neurons of the substantia nigra. But the extent of loss appears to vary from person to person.
And when we say ‘loss’, the situation is not black and white. It is not a case of one day there is a dopamine neuron and the next there isn’t. There appears to be a variable and gradual process, which may leave cells – that were formerly known as ‘dopamine neurons’ – in different states of being.
Huh? What does that mean?
There is research suggesting that as dopamine neurons are being lost in Parkinson’s, they lose their ability to produce dopamine before they actually degenerate. They shut down the dopamine production and enter a ‘dormant’-like status.
Which brings us to the research report we are going to review in today’s post.
Recently some Korean researchers published a report which explores this idea of ‘dopamine neuron loss’.
This is the study:
 Title: Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease.
Title: Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease.
Authors: Heo JY, Nam MH, Yoon HH, Kim J, Hwang YJ, Won W, Woo DH, Lee JA, Park HJ, Jo S, Lee MJ, Kim S, Shim JE, Jang DP, Kim KI, Huh SH, Jeong JY, Kowall NW, Lee J, Im H, Park JH, Jang BK, Park KD, Lee HJ, Shin H, Cho IJ, Hwang EM, Kim Y, Kim HY, Oh SJ, Lee SE, Paek SH, Yoon JH, Jin BK, Kweon GR, Shim I, Hwang O, Ryu H, Jeon SR, Lee CJ.
Journal: Current Biology. 2020 Jan 7. [Epub ahead of print]
PMID: 31928877
In this study, the researchers were interested in the reversible nature of some medical conditions which have “Parkinson’s-like” features.
Wait. What? There are reversible forms of Parkinson’s?
No. But there are certain medical conditions which have “Parkinson’s-like features” that can be corrected over time.
For example, tremors and other Parkinson’s-like movement features are associated with vitamin deficiency, particularly for vitamins B1, B6 and especially B12. Shakiness and tremors can occur in even mild B12 deficiency. But by correcting these deficiencies, the “Parkinson’s-like features” disappear.

Infections of Epstein‐Barr virus have also been reported to cause transcient Parkinson’s-like features (Click here to read about an example this).
The Parkinson’s-like features associated with these conditions appear to be brought on by a temporary depletion of dopamine in the brain. The dopamine neurons appear to be shutting down their primary function of dopamine production while they deal with the particular infection or stress-inducing event, and then they re-start production once the problem has been resolved.
The Korean researchers were intrigued by the idea that this dopamine depletion is transcient or reversible in these situations, and they wanted to explore the mechanisms of it further.
They hypothesised that dopamine neurons in substantia nigra may lose their ‘dopamine producing’ ability and become dormant when the dopamine neuron activity is suppressed. That is to say,they were wondering if continuous inhibition of dopamine neuron activity could be a mechanism which would result in the cells stopping dopamine production.
And the researchers investigated this idea by inhibiting dopamine neuron activty using optogenetics.
What is optogentics?
Optogenetics is a wonderous technology that involves using light to activate or inhibit cellular activity (Click here to read a previous SoPD post on this topic).
Watch this video to learn more about optogenetics:
By introducing the DNA of light sensative ion channels into dopamine neurons, the researchers could control the activity of the cells using light, and they inhibited them completely.
Their initial experiments found that chronic inhibition of the activity of substantia nigra dopamine neurons in rats caused long-lasting reductions in TH protein levels, leading to parkinsonian motor symptoms.
Hang on a second. What is TH protein?
Tyrosine hydroxylase (or TH) is an enzyme involved in the production of dopamine. The basic rule is: If there is no TH, there is no dopamine.
There are two key enzymes involved in dopamine production:
- Tyrosine hydroxylase (or TH)
- Aromatic L-Amino Acid Decarboxylase (or AADC)
Dopamine is generated from tyrosine, which is absorbed from the blood. The tyrosine is firstly altered by TH, and it becomes L-DOPA (the same protein that is used as a treatment to alleviate Parkinson’s symptoms). And then L-DOPA is converted into dopamine by an enzyme called Aromatic L-Amino Acid Decarboxylase (or AADC).
 Source: Quora
Source: Quora
TH is considered a “rate-limiting” enzyme as the levels of TH are influenced and tightly regulated by numerous factors. In this fashion, TH can limit the rate of dopamine production based on what is needed and on what is happening to the cell.
AADC, on the other hand, is not rate limiting and it is more free. It is allowed to do what it wants.
When dopamine neurons were continuously inhibited (by optogenetics), the researchers found that levels of TH dropped dramatically. But curiously, there was no distinct neuronal cell loss.
The dopamine neurons simply stopped producing TH protein, but continued to hang around… just chilling I guess. They become ‘dormant’.
|
RECAP #2: Some medical conditions are associated with ‘Parkinson’s-like features’ (for example, tremor or gait issues), which are transcient and reversible. Dopamine neurons can become ‘dormant’ when inhibited for long periods of time, by shutting down their dopamine producing machinery.
|
How did the researchers know that the cells were still there?
Because they could see them using a particular staining technique that allowed the researchers to visualise the TH-negative, but AADC-positive cells in the substantia nigra.
Cool.
Indeed.
And an even cooler part of the research came next: The researchers found that in three different models of Parkinson’s (MPTP, 6-OHDA, & A53T-transgenic mice) there was evidence of TH negative/AADC positive cells in the substantia nigra.
Cool encore.
Ah, but wait there’s more.
The investigators reported that chronic activation (using optogenetics again) of these TH negative/AADC positive cells in the substantia nigra in rat models of Parkinson’s was enough to reduce some of the behavioural symptoms via the recovery of TH protein in the neurons that had lost their TH protein.
The researchers concluded that their results “suggested that suppressing or activating the substantia nigra neuronal activity is sufficient to cause or alleviate parkinsonian motor symptoms, respectively“.
Interesting. So summing up? What does it all mean?
No, wait. We’re not there yet.
This story has a long way to go.
The researchers wanted to try and put these findings into context with regards to Parkinson’s, so they next shifted their attention to MAO-B.
What are MAO-B?
Monoamine oxidase B (or simply MAO-B) is the first enzyme in recycling old dopamine. It breaks dopamine down into DOPAL.
 The recycling of old dopamine. Source: Google.sr
The recycling of old dopamine. Source: Google.sr
By blocking MAO-B with specific inhibitors (like rasagiline), neurologists can increase levels of dopmaine in the brains of people with Parkinson’s.
How could MAO-B help with dormant dopamine neurons?
Well, MAO-B doesn’t just increase dopamine levels.
It is also involved with the production of GABA.
What is GABA?
Gamma-Aminobutyric acid (or GABA) is a chemical in the brain that is used for transmitting a signal between neurons. Hence, it is called a ‘neurotransmitter’ – similar to dopamine (discussed above). The signal GABA usually passes on is inhibitory, reducing the chance that the recieving cell will be activated.
In 2014, the Korean researchers published this report:
 Title: Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition.
Title: Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition.
Authors: Yoon BE, Woo J, Chun YE, Chun H, Jo S, Bae JY, An H, Min JO, Oh SJ, Han KS, Kim HY, Kim T, Kim YS, Bae YC, Lee CJ.
Journal: J Physiol. 2014 Nov 15;592(22):4951-68.
PMID: 25239459 (This report is OPEN ACCESS if you would like to read it)
In this study, the investigators discovered that astrocytes produce and release the neurotransmitter GABA, but they do it via a mechanism involving MAO-B.
Astrocytes? Can you remind me what they are?
Astrocytes are ‘helper cells’ in the brain. They provide nutrients to neurons and make sure the environment surrounding the neurons is balanced and supportive.
 An astrocyte. Source: Wikipedia
An astrocyte. Source: Wikipedia
The behaviour of astrocytes is very important to the wellbeing of neurons.
Astrocytes have two basic states: activated and non-activated.
When all is well in the brain, astrocytes are at peace and going about their usual business of monitoring the surrounding area and making sure that all of the neurons are ok and well nurtured. But when stress or problems arise, astrocytes will become activated and change their behaviour. They go from nice, caring little cells to something completely opposite: when trouble kicks off, activated astrocytes become utterly ruthless in order to preserve the greater good.
The Korean researchers discovered that astrocytes produce GABA (via MAO-B) especially when they become reactive under pathological conditions. It may be a mechanism of better controlling the environment around them (by limiting neuronal activty) as they try to regain control during periods of cellular stress.
|
RECAP #3: MAO-B is a protein involved in breaking down excess dopamine. Neurologists use drugs inhibit MAO-B from doing its job to allow for more dopamine to be available in the brains of people with Parkinson’s. These drugs are called MAO-B inhibitors. MAO-B is also involved with the production of GABA in helper cells (astrocytes) in the brain. GABA is an inhibitory neurotransmitter. The production of GABA by astrocytes increases in situations of pathology or stress, and may be occuring in Parkinson’s.
|
Thus, the researchers were interested in testing whether GABA released by astrocytes near dopamine neurons might be inhibiting their activity and increasing the chance that they become dormant.
And what did they find?
Firstly, the researchers found that mice with no MAO-B, but which produced high levels of alpha synuclein (A53T α-syn) were relatively resistant to alpha synuclein-induced dopamine cell loss in the substantia nigra (compared to normal mice).
Next, they reported that blocking the production of GABA in astoryctes replicated the therapeutic effect of optogenetic activation.
And third, they demonstrated that direct injection of pharmacological agents (which manipulate GABA levels) into the substantia nigra could reduce levels of TH in dopamine neurons, resulting in the appearance of Parkinson’s-like features.
Thus, the researchers proposed that GABA, synthesized by MAO-B in astrocytes, may be being released and accumulating in the extracellular space of the substantia nigra. And this action can inhibit dopamine neurons, reducing their levels of TH and resulting in motor impairments similar to those seen in Parkinson’s.
Interesting. Does this result mean that MAO-B inhibitors like rasagiline could be neuroprotective?
So this is a very controversial topic in the field of Parkinson’s research. It’s a big deal that divides investigators.
It is fair to say that the answer is not clear.
There have been two major clinical trials to try and determine whether the MAO-D inhibitor rasagiline has any disease-modifying effects.
The first study was called the “TVP-1012 in Early Monotherapy for PD Outpatients” (or TEMPO) study, and the results of that study were published in 2004:
 Title: A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease.
Title: A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease.
Authors: Parkinson Study Group
Journal: Arch Neurol. 2004 Apr;61(4):561-6.
PMID: 15096406 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators conducted a randomised double-blind (meaning that no one knew who was getting what treatment), parallel-group, delayed-start clinical trial. The ‘parallel-group, delayed start’ design involved participants being randomly assigned to receive rasagiline (either 1 or 2 mg per day) for 1 year OR to receive placebo for 6 months followed by rasagiline (2 mg per day) for 6 months. These two groups being treated differently in parallel could then be compared at the end of the study to determine if the drug had any disease modifying effect – if there is an effect, the group that received rasagiline for the full 12 month should be better off than the group that had placebo for 6 months before being given rasagiline for 6 months (as a result of being treated with the drug for a longer period).
A total of 404 people with recently diagnosed Parkinson’s that was not being treated with dopamine-based therapy (for example L-dopa) were recruited at 32 different research sites across the United States and Canada. After the 12 month long study, the researchers found that the subjects receiving either dose of rasagiline (1 or 2 mg per day) for the whole year were better off by approximately 2 points on their Unified Parkinson’s Disease Rating Scale (UPDRS) scores when compared to the group that received the placebo plus rasagiline. This result suggests that there was an positive effect from taking the MAO-B inhibitor.
It was a minimal effect, but an effect nonetheless.
The second large clinical study that has looked at this issue was called the “Attenuation of Disease Progression with Azilect Given Once-daily” (or ADAGIO) study (Azilect being the commercial name for rasagiline):
 Title: A double-blind, delayed-start trial of rasagiline in Parkinson’s disease.
Title: A double-blind, delayed-start trial of rasagiline in Parkinson’s disease.
Authors: Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F, Tolosa E; ADAGIO Study Investigators.
Journal: N Engl J Med. 2009 Sep 24;361(13):1268-78.
PMID: 19776408 (This article is OPEN ACCESS if you would like to read it)
This second study followed a similar design to the first study with a total of 1176 people with untreated Parkinson’s being randomly assigned to either receive rasagiline (at a dose of either 1 mg or 2 mg per day) for 72 weeks OR receive a placebo treatment for 36 weeks followed by rasagiline (at a dose of either 1 mg or 2 mg per day) for 36 weeks.
The results of the ADAGIO study were not as clear as the TEMPO trial. Only the group of subjects receiving 1 mg daily for the full study had significantly better UPDRS scores at the end of the study. The group receiving the higher dose of rasagiline (2 mg per day) for the entire study showed no different to the group that received the placebo treatment for the first 9 months of the study. The investigators concluded that rasagiline could possibly be having a disease-modifying effect in the 1 mg group.
An initial interpretation of this data could be that there isn’t much evidence for a major disease modifying effect in the clinic for rasagiline in Parkinson’s.
This conclusion is confounded, however, by a 6.5 year follow up study of the TEMPO trial participants. That follow up study found that the participants that were in the ‘rasagiline for the whole trial’ group were 16% better off than the participants in the placebo-rasagiline treated group, even at 6 years after the trial was finished (Click here to read more about this follow up study).
So you can see why this issue is so debated by member of the Parkinson’s research community (Click here for an interesting OPEN ACCESS review of the MAO-B inhibitor clinical trials).
|
RECAP #4: The Korean researchers reported that GABA, produced by MAO-B in astrocytes, may be being released and accumulating in the extracellular space of the substantia nigra. And this action can inhibit dopamine neurons, reducing their levels of TH, and resulting in motor impairments similar to those seen in Parkinson’s. MAO-B inhibitors have been clinically tested to assess their potential effect in disease modifying studies, but the results have not been conclusive.
|
Interesing. So now is it time to sum up? What does it all mean?
Nope, not just yet.
The Korean researchers next wanted to look in postmortem brains from people with Parkinson’s to see if there were any TH-negative, AADC-positive ‘dormant dopamine’ neurons present.
And what did they find?
They found that the number of AADC-positive neurons as well as the number of TH-positive neurons was significantly decreased according to the severity of the disease, suggesting that (as we discussed above) there is dopamine neuron cell death in Parkinson’s.
BUT, in both mild and severe cases of Parkinson’s, the portion of remaining AADC-positive neurons in substantia nigra was greater than those of TH-positive neurons, which suggests that existence of dormant dopamine neurons.
In fact, in cases of mild severity, they found 54.7% of AADC-positive cells were TH-negative, and in more severe cases this portion was 27.0%. This suggests that even in cases of severe Parkinson’s they may be dormant cells.
Wow. Has anyone else ever reported this ‘dormant dopamine neuron’ finding before?
Yes, it has been observed.
In 2013, for example, this report was published:
 Title: Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease.
Title: Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease.
Authors: Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT.
Journal: Brain, 2013 Aug;136(Pt 8):2419-31.
PMID: 23884810 (This report is OPEN ACCESS if you would like to read it)
In this study, the investigators analysed the postmortem brains of 28 people with Parkinson’s (and compared them with 9 control brains). The post-diagnosis interval (that is the time between diagnosis of Parkinson’s and death) ranged from 1–27 years.
In the substantia nigra, the researchers reported that there was a 50–90% loss of TH-positive neurons from the earliest time points studied, with less than 20% cell loss thereafter. This suggests that a lot of the TH-postitive dopamine neurons are “lost” before diagnosis.
BUT, when the researchers counted the number of neuromelanin cells in the substantia nigra, however, there was only a small reduction (~10%) in the number of cells in the one case that they evaluated 1 year post-diagnosis. And there was a variable (30 to 60%) loss over the first few years post-diagnosis, with more gradual and subtle loss in the second decade.
And the researchers were very clear about this – “at all time points, there were more melanin-containing than tyrosine hydroxylase-positive cells“.
And they concluded their study by suggesting that “the number of tyrosine hydroxylase-positive neurons is less than the number of melanized neurons at all time points, suggesting that some nerve cells are likely to be damaged but to have not yet degenerated“.
And then in thediscussion, they propose that “the persistence of populations of melanin containing neurons in comparison to the number of tyrosine hydroxylase-immunoreactive neurons for decades after diagnosis, suggests that trophic or regenerative therapies might still have value even in the later stages of the illness“.
So now can I ask what does it all mean?
The paper reviewed in today’s post fascinated me for various reasons, but I was a rather reluctant to write a post about it. I did not want folks to get excited and start chatting on the forums about how the dopamine cells aren’t all gone, and they are just ‘dormant’. The data suggests that the loss of dopamine neurons is a very slow process, but cells do appear to be disappearing over time.
But I decided to write a post because there is an increasing amount of research looking at this area and it is important for us to discuss these sorts of findings. The obvious BIG question is whether those cells can come back from that dormant state of purgatory, and this is yet to be determined in humans.
But before that question can even be addressed, we require a better understanding of the nature of the ‘dormant’ cells. What exactly are they doing? There have been recent reports of ‘senescent cells’ in the brain, and how some of them could be having a negative effect on the surrounding environment (Click here to read a previous SoPD post about this). Could these dormant dopamine neurons be having a negative effect?
I have to admit that I am intrigued by the finding of more neuromelanin-positive (or AADC-positive) cells in the Parkinsonian substantia nigra than TH-positive cells. But there is still a lot to be learnt here before we can start asking if they are potentially retrievable.

All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from Thesaurus

Thanks as always Simon. I need to read this one a couple times… my standing thought l grasping for hope scenario:
– If half of DA neurons are ‘dead’ at symptom offset
– if 20% can be rescued with a neurprotective + disease modifying therapy
– does that mean early disease can reversed by getting back to 60% capacity?
More importantly, I think you missed an opportunity for for a “Princess Bride mostly dead Miracle Max” reference
LikeLike
Simon,
Very interesting. I hope your post is discussed in the forums.
Any theory about the survival, or otherwise, of dopamine producing cells must be able to explain these observations why, 14 years post-diagnosis, can I still:
– walk near normally 12 hours after my last dose of levodopa (half life about 90 minutes), and 24 hours after my last doses of ropinirole (6 hours) and rasagiline;
– do well on the side-to-side tap test score (30 seconds each hand, left and right scores added together) where my score goes from approximately 30 to 60 as I go from maximally “off” to maximally “on” following taking a dose.
John
LikeLike
There may be a mechanism for GABA-inhibition of Ca spiking impairing neuronal survival:
.https://www.frontiersin.org/articles/10.3389/fnins.2018.00819/full
(” is increased cytosolic calcium always bad…”)
Maybe MAO-B inhibitors exert and unrecognised protective effect by this route: lowering GABA> restoring Ca spiking> reviving nuclear Ca-sensiitve transcription.
Is there a dosage regime for say rasagiline than can depress GABA without addng to the DOPAL load ?
LikeLike
Slightly off topic.
Do meds that increase gaba make PD worse? Or better?
“In man, gabapentin has been demonstrated to increase GABA concentrations.”
“While the exact cause of chronic neurodegeneration of PD is not known, increasing evidence suggests that chronic inflammation is the fundamental process mediating the progressive nature of the neurodegeneration characteristic of PD (Glass et al., 2010). Induced by GABA deficit, inflammation may amplify the pathology. ” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4899466/
LikeLike
Hi Simon, first, I wanted to thank you for putting so much effort into this site that I recommend to my patients with PD regularly. Even though I try to keep up on much of the PD literature, I am always learning new information from your blogs. I wanted to make you aware of another study (https://www.ncbi.nlm.nih.gov/pubmed/23152586) that my colleagues an I performed with brain tissue from the Honolulu Asian Aging Study wherein we demonstrated the presence of TH negative pigmented nigral neurons in cases of incidental Lewy body disease, without clinical Parkinsonism. Perhaps most interestingly, we also showed the presence of these dysfunctional nigral neurons in cases of incidental Lewy body pathology that did not have evidence of Lewy pathology in the substantia nigra, suggesting the nigral neuron dysfunction and death can occur independent of alpha-synuclein aggregation. Warm regards, and keep up the good work! John Duda, MD
LikeLike
To do a little back of the envelope math here:
If there is say 70 percent (“50-90%”) loss of TH-positive neurons from the earliest time points studied (and more minor and slow loss thereafter), and say 45% (“30 to 60%”) loss of neuromelanin-containing neurons over the first few years post-diagnosis, then after those first few years of diagnosis there should remain about 25 percent of the original neurons that are impaired but not yet dead (plus another 30 percent that are still fully functional).
So *if* those neurons could somehow be revived, then considering that symptoms do not appear until neurons are reduced very substantially, adding back 25 percent of the original neurons could have a very dramatic effect on symptoms.
But even without such a revival, if 25 percent of original neurons were in this dormant condition, and if the main aspect of their dormancy were just the cessation of their TH production, then their axon terminals would still be available to import (via the dopamine transporter) dopamine created by hijacked 5-HT neurons from exogenous levodopa. And that would have a very significant effect upon the efficacy of levodopa therapy, because those neurons would be able to fire and disperse the dopamine that they had imported, and since the axon terminals can store about 2.5 times (I think I saw that number in a rodent study) the amount of dopamine produced by a dopamine neuron itself under normal physiological conditions, that would provide a very significant buffer for dopamine from exogenous levodopa, which would insulate the patient from the ups and downs of the medication cycle, and would also result in the release of dopamine under control of those neurons, based on the usual criteria for deciding whether such release was needed at any given moment.
So even if there is no way to revive those neurons, might they not be playing a very important role already in maintaining the effectiveness of levodopa therapy? If so, then simply keeping them from fully degenerating to the point that they are fully dead could be a valuable goal.
Now, are these damaged neurons in effect “senescent” as you suggest might be the case? And are they then sending out constant signals to rally the immune system to kill them off, which adds to inflammation?
I would suspect that it is not quite the same thing, since senescent cells decline due to age, while these neurons are probably the victims of both aggregated alpha-synuclein and an active neuroinflammatory autoimmune process that is a central aspect of Parkinson’s disease. But certainly having damaged cells around could lead to the production of Damage-Associated Molecular Patterns, which could encourage autoimmune inflammation, leading to additional damage in the surrounding area.
Since these damaged neurons are, at present, irreplaceable, and may (as I’ve speculated above) still be performing a useful function, it seems to me that an approach that protects them by trying to calm the immune system using anti-inflammatories (e.g., Longvida curcumin, baicalin, and EGCg) and antioxidants (e.g., NAC and, once again, EGCg), rather than trying to kill them off (as in some approaches to cell senescence that use fasting or fisetin to allow senescent cells to be successfully destroyed) could be the most helpful approach.
Also, if there is to be any hope of reviving these cells, I would think that this would require reducing the damaging processes that impaired them in the first place. And once again, I would suspect that anti-inflammatory and antioxidant therapies could hold part of the key to providing these cells a more favorable environment for recovery. That, and shoring up mitochondrial function (e.g., through ubiquinol supplementation).
LikeLike