![]()
|
Moving forward into 2019 and beyond, we are going to be getting more sophisticated and targetted with our clinical trials for Parkinson’s. We are gradually moving away from the days when a drug was tested on anyone in the Parkinson’s-affected community, and heading for an age of sub-type specific treatments (Click here for a previous SoPD post on subtyping efforts for PD). As part of this shift, there are a series of ongoing studies that are trying to identify not only the clinical & biological characteristics of those Parkinson’s sub-types, but also individuals who may already be in those groupings. One such study is called “Rapsodi” – and it is focused on the identification of people with a particular genetic risk factor of PD – the GBA gene – who also demonstrate the early signs of Parkinson’s. In today’s post, we will discuss what GBA is, how it is associated with Parkinson’s, and why the Rapsodi study is worthy of the PD community’s attention.
|

Ambroxol. Source: Skinflint
The clinical trial of Ambroxol in Parkinson’s that has been conducted in London (UK) is close to announcing their final results. The Ambroxol study report should be published in early 2019.
What is the ambroxol study?
Started in February 2017, the Ambroxol study (named AiM-PD – Ambroxol in Disease Modification in Parkinson Disease) is a phase IIA prospective, single-centre, open label clinical trial to evaluate the safety, tolerability and pharmacodynamic effects of Ambroxol in Parkinson’s (Click here to read more about this trial and click here for the press release announcing the start of the study).
This trial, which is funded by the Cure Parkinson’s Trust and the Van Andel Research Institute (USA), has been conducted at the Royal Free Hospital in London (UK). The study has involved 20 people with Parkinson’s self-administering Ambroxol (in 60 mg per tablet) over a 6 month time frame. The participants were given 5 escalating doses of the drug for the first few weeks of the study (from 60 mg three times per day, gradually building up to 420 mg three times a day after the first month of the study).
But hang on a second. What is exactly is Ambroxol?
Ambroxol is a commonly used treatment for respiratory diseases (the respiratory system being the lungs and related components required for breathing). Ambroxol promotes the clearance of mucus and eases coughing. It also has anti-inflammatory properties, reducing redness in a sore throat. It is the active ingredient of products like Mucosolvan, Mucobrox, and Mucol.
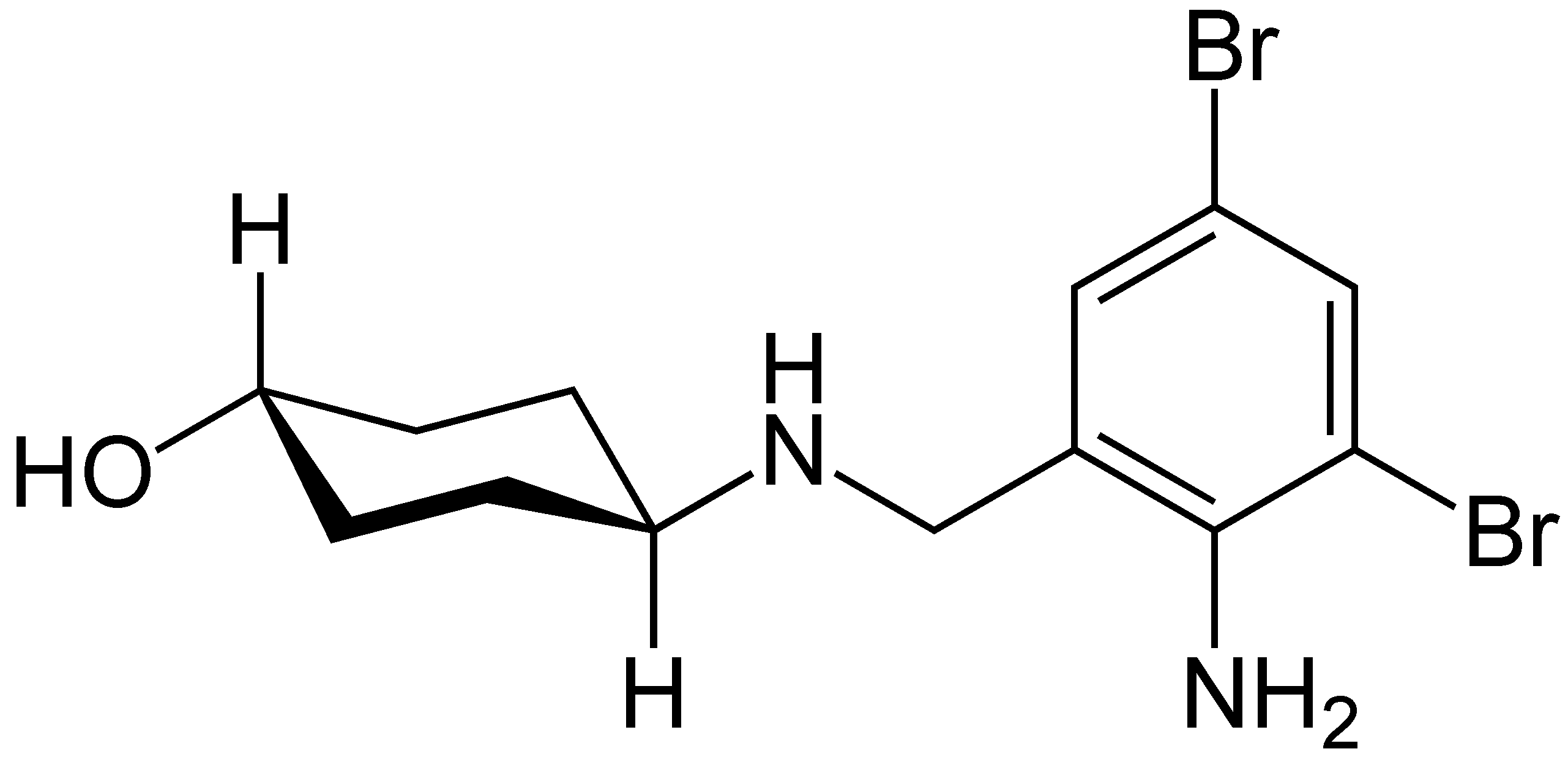 Ambroxol. Source: Wikipedia
Ambroxol. Source: Wikipedia
Researchers believe that Ambroxol could help with Parkinson’s in two ways:
1. Ambroxol is believed to triggers exocytosis of lysosomes (Source). Exocytosis is the process by which waste is exported out of the cell (the opposite of ‘endocytosis’ which we discussed above). Lysosomes are the bags of digestive that rubbish and waste is put into inside a cell for recycling. By encouraging lysosomes to undergo exocytosis and spit their contents out of the cell – digested or not – Ambroxol allows the cell to remove waste effectively and therefore function in a more normal fashion.

Exocytosis. Source: Socratic
2. Ambroxol has been shown to increase levels of the enzyme glucocerebrosidase in the brain (Source).
Thus, by administering Ambroxol to people with Parkinson’s, researchers are hoping to raise levels of the enzyme glucocerebrosidase to help digest proteins and increase the excretion of this waste from cells. This would ideally keep cells healthier for longer and slow down the progression of Parkinson’s.
And what exactly is glucocerebrosidase?
Glucocerebrosidase (also known as GCase) helps with the digestion and recycling of various proteins (particularly glucocerebrosides) inside cells. The enzyme is located and active inside small bags of degradative enzymes – these bags are the lysosomes I mentioned above – that can be found inside of cells.
How do these lysosomes work?
On a continual basis, small parts of the external layer of the cell membrane is brought inside the cell. This is a process called endocytosis. It occurs when the cell consumes resources from the outside world in order to find what it needs to function and survive. As a section of cell membrane is brought into the cell, it forms a vesicle (which is a term used to refer to small spherical bags of stuff inside cells). Given the process by which that vesicles was formed, it is referred to as an endosome (sometimes it is also called a vacuole).
 Source: Socratic
Source: Socratic
Once the endosome is inside the cell and detached from the rest of the membrane, it will bind to another vesicle which is called a lysosome. And as I mentioned above, lysosome is a small bag that is full of digestive enzymes, which help to break down the contents of the endosome.

How lysosomes work. Source: Prezi
The lysosome will fuse with the endosome/vacuole and the enzymes from the lysosome will mix with the material in the vacuole and digest it (or it break down into more manageable components).
This enzymatic process works in a very similar fashion to the commercial products that you use for washing your clothes.
 Enzymatic degradation. Source: Samvirke
Enzymatic degradation. Source: Samvirke
The reagents that you put into the washing machine with your clothes contain a multitude of enzymes, which help to break down the dirty, bacteria, flakes of skin, etc that cling to your clothes. Each enzyme breaks down a particular protein, fat or such like. And this is very similar to the collection of enzymes in the lysosome. All of them are needed to break down all of the contents of the endosome.
And if one of those enzymes – such as glucocerebrosidase – is faulty (due to a genetic mutation), then the enzymatic process is disrupted, which could result in the build up of un-degraded material over time.
Ok, but why would Ambroxol possibly help people with Parkinson’s?
The enzyme Glucocerebrosidase is produced by cells using instructions provided by the GBA gene (a gene is a section of DNA that provides the instructions for making a particular protein).
Genetic variations in the GBA gene are associated with two conditions:
- Parkinson’s
- Gaucher disease.
What is Gaucher disease?
Gaucher (pronounced “go-shay”) disease is a rare inherited genetic disorder characterised by the build up in cells of a fatty chemical called glucocerebroside. Because the body cannot break down this chemical, swollen fat-laden cells build up in certain areas of the body, such as the spleen, liver and bone marrow. These cells are referred to as ‘Gaucher cells’.
 Swollen Gaucher cells (circled in red). Source: Imagebank
Swollen Gaucher cells (circled in red). Source: Imagebank
The disorder results from the deficiency of the enzyme glucocerebrosidase, which usually breaks down glucocerebroside. The incidence of Gaucher disease is about one in every 40,000 live births (Source), and the condition manifests itself in several way, from reduced bone density to swollen liver and spleen:
 The signs of Gaucher disease. Source: Rare2aware
The signs of Gaucher disease. Source: Rare2aware
What happens to the cells in Gaucher disease?
Macrophage are one type of cell that is particularly affected in Gaucher disease. They are a type of blood cell that is responsible for detecting, engulfing and destroying dangerous pathogens and apoptotic cells. Below is a schematic of a macrophage, consuming orange pathogens (left), digesting them, and releasing the waste (on the right):
 A schematic of a macrophage. Source: Meducator
A schematic of a macrophage. Source: Meducator
Macrophage travel the body, swallowing anything that they don’t like the look of. In order to break down everything it swallows, a macrophage must have a full complement of digestive enzymes. But – as you can see in the image below – without glucocerebrosidase, the macrophage has trouble digesting fatty chemicals like glucocerebrosides and the lysosomes start to accumulate in the cell, causing the cell to swell up.
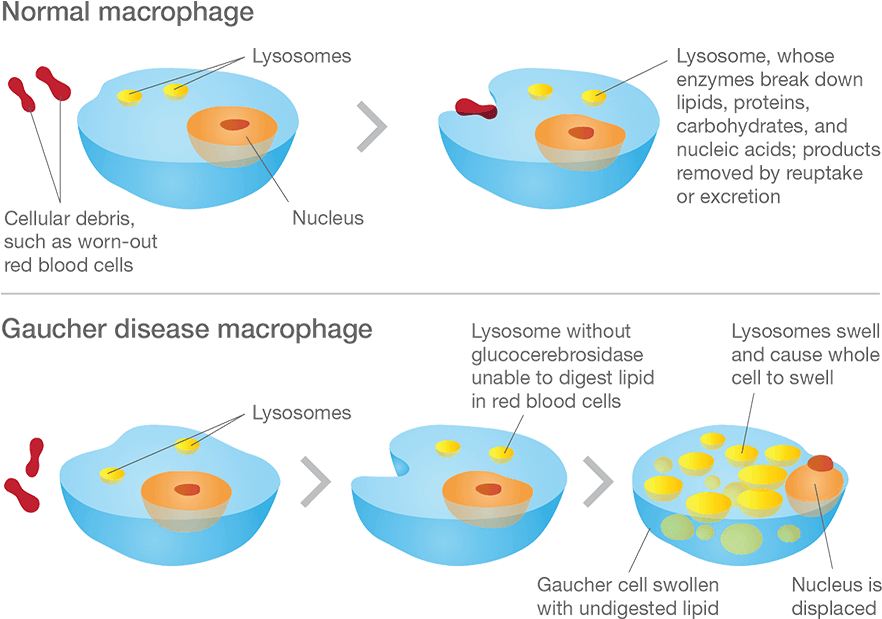 Gaucher disease. Source: Gaucherawareness
Gaucher disease. Source: Gaucherawareness
Are there different types of Gaucher disease?
There are three types of Gaucher disease, and they are all caused by genetic mutations in the GBA gene:
- Type I – (also called the “non-neuropathic” type) this is the most common; having said that it mainly occurs in Ashkenazi Jews (x100 more than the general population). The median age at diagnosis is 28 years of age, and life expectancy is only mildly decreased. As the “non-neuropathic” label suggests, there are no neurological symptoms.
- Type II – is characterised by neurological problems in small children. The glucocerebrosidase enzyme is barely present in the lysosomes. Prognosis is poor (death before the age of three).
- Type III – (the Swedish variety) occurs in people from the Norrbotten region in Sweden. This group develops the disease somewhat later, but most most do not survive their 30th birthday.
There are three subtypes of Gaucher disease, which are characterised by the presence or absence of neurological issues, and by the rate of disease progression and severity:
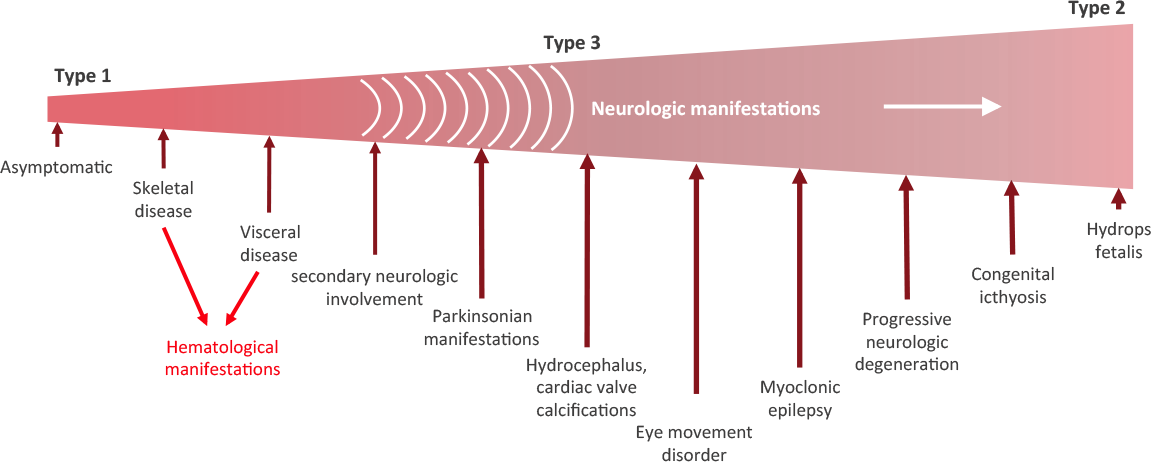 Stages of Gaucher disease. Source: Gaucherawareness
Stages of Gaucher disease. Source: Gaucherawareness
You said Gaucher disease is inherited. So if one of my parents has Gaucher disease will it affect me?
As I mentioned above Gaucher disease is an inherited genetic disorder, caused by mutations in the GBA gene. Humans normally have two copies of the GBA gene. If one copy of the GBA gene is faulty due to a genetic mutation, the person will not develop Gaucher’s disease, because the one remaining functional gene will be able to produce enough of the glucocerebrosidase enzyme.
Gaucher disease is considered an autosomal recessive disorder. This means that two copies of an abnormal gene must be present in the individual in order for the disease to develop. A person with just one faulty gene will not get sick, but they will be a carrier. To develop Gaucher disease, you need to have two genetic mutations in the GBA gene – one from your mother and one from your father.
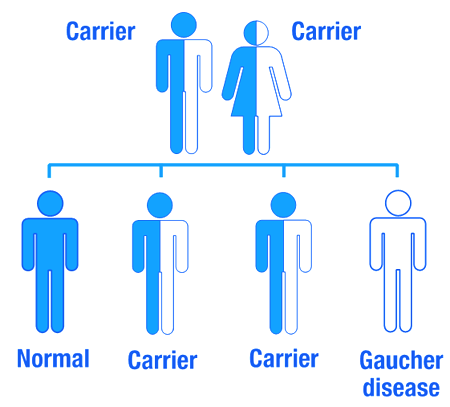 An autosomal recessive disorder. Source: Myhealthyfeeling
An autosomal recessive disorder. Source: Myhealthyfeeling
So even if one of your parents has been diagnosed with Gaucher disease, you will not necessarily develop it if the other parent does not have a mutation in their GBA gene.
Ok. All of this is interesting, but how is Gaucher disease associated with Parkinson’s?
In the 1990s, physicians began to notice patients with both Gaucher and Parkinson’s. An example of this was a report published in 1996 that described six people with Gaucher disease who also exhibited an early-onset, severe form of Parkinson’s with cognitive decline:
 Title: Occurrence of Parkinson’s syndrome in type I Gaucher disease.
Title: Occurrence of Parkinson’s syndrome in type I Gaucher disease.
Authors: Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A.
Journal: QJM. 1996 Sep;89(9):691-4.
PMID: 8917744 (This article is OPEN ACCESS if you would like to read it)
In this study, the Israeli researchers report on 6 people with Type I Gaucher disease (which up until that point had not been considered neuronopathic). All six of the subjects also exhibited the hallmark of a rather severe form of Parkinson’s, which made its appearance in the 4th to 6th decade of life and displayed an aggressive progression and was largely unresponsive to conventional anti-Parkinson therapy (such as L-dopa).
These initial reports were followed by many additional studies which eventually started pointing towards the GBA gene as the likely risk factor for this form of Parkinson’s, including this study:
 Title: Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews.
Title: Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews.
Authors: Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R.
Journal: N Engl J Med. 2004 Nov 4;351(19):1972-7.
PMID: 15525722 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators examined 99 individuals with idiopathic Parkinson’s from an Ashkenazi Jewish background. Thirty-one of them (31.3%) had one or two mutations in their GBA gene. And of all the individuals with Parkinson’s, the subjects who were carriers of GBA mutations were younger than those who were not carriers (mean age at onset being 60 years vs. 64 years).
And this result is similar to what has been seen in larger follow up studies (Click here for an example).
So genetic variations in the GBA gene are associated with Parkinson’s?
Yes. It is one of the main genetic risk factors for the condition.
It is now believed that approximately 5%–8% of people with Parkinson’s have a genetic mutation in the GBA gene (Click here and here to read more about this). According to the Michael J Fox foundation webpage on GBA “up to 10 percent of people with PD in the United States carry” a genetic variation in the GBA gene.
What do we know about the GBA gene?
GBA is a very large gene and there are numerous genetic variants spread across its length. The most common mutations are located in positions N370S and L444P. These mutations cause a reduction in the enzymatic activity of the glucocerebrosidase enzyme.
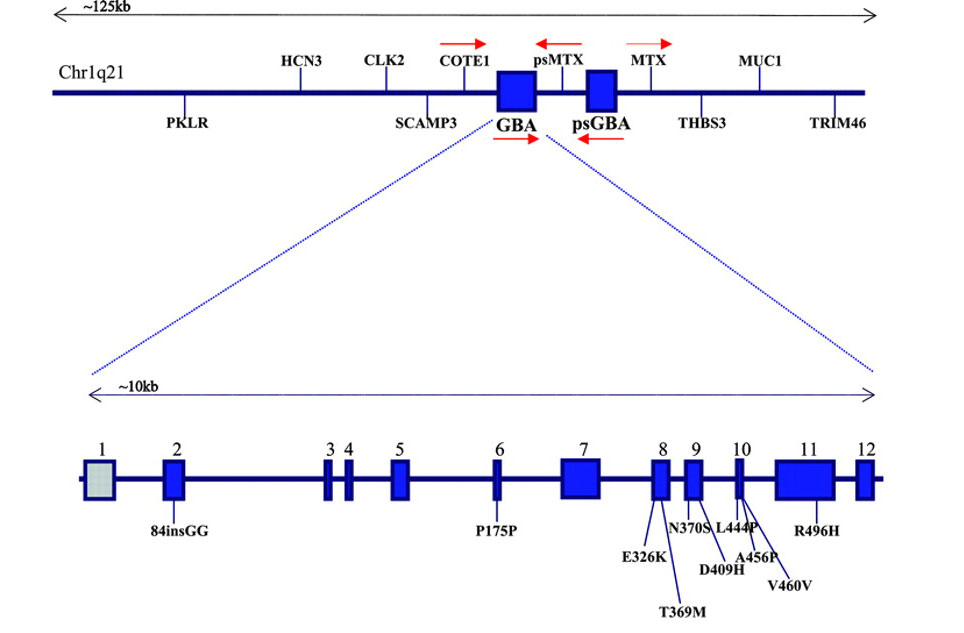 The GBA gene and mutations associated with Parkinson’s. Source: Neurology
The GBA gene and mutations associated with Parkinson’s. Source: Neurology
And as I mentioned above generally, people with GBA-associated Parkinson’s will exhibit more severe symptoms than people without a GBA mutation, and this has recently been confirmed in a large clinical observation study:
 Title: Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study.
Title: Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study.
Authors: Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, Bajaj N, Barker RA, Burn DJ, Foltynie T, Hardy J, Wood NW, Ben-Shlomo Y, Williams NW, Grosset DG, Morris HR; PRoBaND clinical consortium.
Journal: J Neurol Neurosurg Psychiatry. 2018 [Epub ahead of print]
PMID: 29378790 (This article is OPEN ACCESS if you would like to read it)
In this multi-research centre study, the investigators studied 1893 people with Parkinson’s. Of these 48 (2.5%) had one copy of a known GBA mutation, 117 (6.2%) had a non-GBA genetic variant that had previously been associated with Parkinson’s (such as mutations in genes like LRRK2 or Alpha Synuclein), and 28 (1.5%) carried genetic variants of unknown significance in the GBA gene. As I mentioned above, onte of the most common GBA mutations associated with Parkinson’s is called ‘L444P‘ and this was also the most common GBA mutation observed in this study.
Individuals with Parkinson’s associated GBA mutations were on average diagnosed 5 years earlier compared with non-carriers. They were also more likely to have postural instability and gait difficulties compared with non-carriers. In addition, they had more progressive forms of Parkinson’s (as determined by more advanced Hoehn and Yahr staging – after adjustment for age – compared with non-carriers).
No differences was observed in cognitive function between GBA mutation carriers and non-carriers. Cognitive impairment/dementia have been reported in other studies at a later stages of the condition (Click here to read more about this). This observation led the researchers to conclude that “this offers an important window of opportunity for potential disease-modifying therapy that may protect against the development of dementia”.
This is really bad, right? What if I have a GBA genetic mutation?
It is important for readers to understand that our understanding of the genetics of Parkinson’s is basic at best and that this aggressive pattern of disease progression in GBA-associated Parkinson’s is not always the case.
In addition, 2-3% of the general population will have a Parkinson’s associated GBA mutation in their DNA, but they never go on to present any of the features of Parkinson’s. And then there are cases of identical twins who both have a GBA mutations, but only one of them has developed Parkinson’s (Click here to read more about this).
Thus, the genetics of Parkinson’s is still very complex, and just because a person has a GBA mutation, it does not necessarily mean that they will go on to develop Parkinson’s or necessarily present the aggressive form of the condition.
More research is required to increase our understanding of this situation.
Which brings us (finally) to Rapsodi.
What is Rapsodi?
Rapsodi stands for Remote Assessment of Parkinsonism Supporting Ongoing Development of Interventions in Gaucher disease.
This is a large internet-based study that aims to identify clues about Parkinson’s many years before any of the movement symptoms associated with the condition start to appear. And the investigators conducting this study are focusing their attention on people with GBA genetic mutations.
Given that only 2-3% of the general population are carrying a GBA mutation, the investigators behind Rapsodi need a very large group of people – who are willing to take part in the study – to register. The large number of participants will allow the researcher to begin to look at patterns of data that are specific to the GBA-associated community as opposed to the GBA un-affected participants.
What does the study involve participants doing?
As they state on the Rapsodi website, it is very straight-forward:
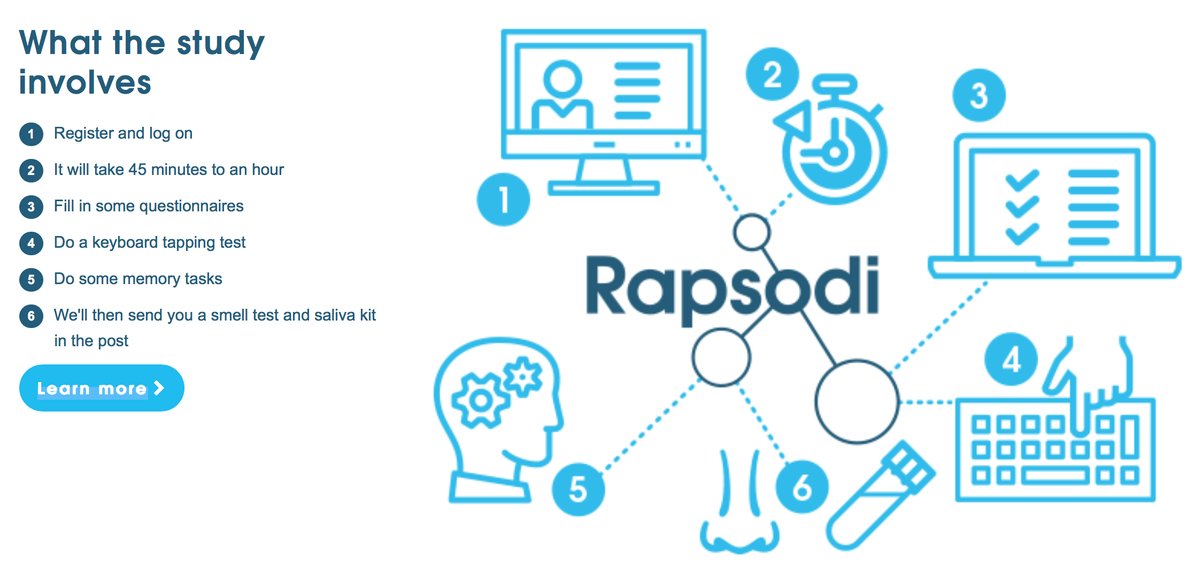
One year later, the study organisers will email you and invite you to re-visit the website (to do questionnaires, keyboard tap tests and memory tasks again). And they may follow up participants in subsequent years.
Who can take part in the study?
The researchers are looking for people who have genetic mutations in the GBA gene, and first degree family members such as parents, siblings and children. They are also seeking spouses of people who carry genetic mutations in the GBA gene. You must be over 18 years of age to take part in the study.
If you are not sure if you have a GBA mutation, you can get involved and find out if you do.
If I get involved in the study will I definitely find out if I have a GBA mutation?
Only if you chose to know.
When you enroll in the study, you will be asked whether you would like to be given the result of a genetic test for the GBA gene (Please note that the researchers will also be looking for mutations in the LRRK2 gene). The decision to know your genetic status will be entirely up to the individual enrolling in the study. If they chose not to, the researchers will keep that information undisclosed.
If you have any questions regarding the Rapsodi study, you can email the coordinators (rapsodi@ucl.ac.uk) and ask them directly.
Why is this study important for the Parkinson’s community?
In addition to telling us a great deal about those who are at risk of developing Parkinson’s, the Rapsodi study will also be able to identify individuals who may wish to be involved with – and could benefit from – future clinical studies looking at GBA-targeting treatments (if they chose to be involved).
This is particularly important if the current Ambroxol study provides results that would justify a large Phase III clinical trial of the drug. Recruitment is one of the main issues that slows clinical development of new therapies, but by identifying the population of interest before even considering the initiation of a trial, the investigators are making life easier for themselves and speeding up clinical trial process.
And a study like Rapsodi could provide participants with options that they would not get if they were not involved.
What sort of options?
Well, the team behind Rapsodi is also coordinating the Ambroxol clinical trial. But in addition to this, they are also working with the biotech company Sanofi Genzyme and conducting a series of clinical trials that are testing a drug that is also focused on individuals with GBA-associated Parkinson’s.
![]()
The ongoing phase II clinical trial, called MOVE-PD, is testing the efficacy and safety of a drug called Venglustat (also known as GZ/SAR402671 – Click here to read more about this clinical trial).
Venglustat is a glucosylceramide synthase inhibitor.
Glucosylceramide synthase is an enzyme involved in the production of glucocerebroside. Thus, by blocking glucosylceramide synthase, Sanofi Genzyme are hoping to reduce the production and consequent accumulation of glucocerebroside in people with a mutation in the GBA gene. This approach is trying to reduce the amount of protein that can not be broken down by the glucocerebrosidase enzyme (Click here to read more on this).
The Ambroxol study and the MOVE-PD study are focused on GBA-associated Parkinson’s. Many individuals in the Parkinson’s community will be unaware that they have a GBA mutation that could make them eligible for taking part in studies like this (if they chose to).
Are there other studies like Rapsodi?
Yes, there are.
We have previously mentioned the PREDICT-PD study here on the SoPD (Click here for one example).
![]()
PREDICT-PD is “a ground-breaking project using simple tests to identify people at high risk of Parkinson’s disease before the symptoms appear“. There is a lot of research suggesting that problems associated with Parkinson’s (such as loss of sense of smell, sleep problems, constipation, anxiety and depression) can begin to appear many years before the movement problems appear and subsequent diagnosis of Parkinson’s is made. If we can better identify individuals at risk of going on to develop Parkinson’s, we may be able to start treating them earlier.
This is the goal of the PREDICT PD study.
They are not particularly focused on a single subtype, but rather trying to identify general factors that may influence risk of developing PD. The PREDICT-PD team have just re-launched their website, it is worth having a look – Click here to learn more about it. The folks at the PREDICT-PD study also maintain a really good blog – Click here to have a look.
In addition to PREDICT-PD, there is also the large Fox Insight study which is sponsored by The Michael J. Fox Foundation.

This online study – with over 29000 participants – invites individuals with Parkinson’s to enrol and every 90 days fill in information relating to life with Parkinson’s. This regular effort provides researchers with a rich database of information. In addition, Fox Insight is a remarkable research platform on which you can even take part in one-time survey studies on topics that specifically interest you.
Definitely worth a look!
So what does it all mean?
Taking part in any kind of clinical research is a very personal choice, and there should never be any cohersion or demand for members of the Parkinson’s community to do anything that they do not want to do. But for individuals who are interested in Parkinson’s research, these observational studies provide an easy “entry-level” way of getting involved. Rather than diving head first into a serious clinical trial, studies like Rapsodi and PREDICT-PD require very little effort from the participants.
And these studies exploring the early stages of Parkinson’s will be absolutely critical as we move ahead.
Going forward you might notice a slight shift in the nature of some of the research being discussed on the SoPD. My focus is still on ‘curative’ approaches to Parkinson’s, but a very hot area of Parkinson’s research at the moment is the pre-motor/prodromal phase of the condition, and we are going to see more and more research coming down the pipe on this topic.
 Source: Guidelinesinpractice
Source: Guidelinesinpractice
The earlier we can tackle Parkinson’s, the better for society in general. That is why a considerable amount of research attention is now being focused on this phase of the condition (with studies like PREDICT-PD and Rapsodi). And the more we can learn about the early stages of Parkinson’s, the more likely we are to identify new biological pathways that can be targeted in future therapies.
As Professor Anthony Schapira (who is co-ordinating the Rapsodi study) says on the Rapsodi website: “every bit of information gets us closer to understanding how to prevent Parkinson’s developing and progressing“.
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from Rapsodistudy

8 thoughts on “Not bohemian, just ‘Rapsodi’”