
|
Recently a really interesting research report was published that presented several rather amazing findings. The researchers forced dopamine-producing cells in a rodent brain to start making a protein called neuromelanin and by doing this, they witnessed the occurence of Parkinson’s-like features (motor issues, Lewy body-like structures, and cell death). The report also suggested a method by which this outcome could be reduced or rescued. But the amazing part is that neuromelanin was previously considered to be protective and this new finding suggests we may need to rethink that idea. In today’s post, we will discuss what neuromelanin is, what this new report found, and how this new knowledge could be useful in the context of Parkinson’s.
|
 Prof Heiko Braak. Source – Memim.com
Prof Heiko Braak. Source – Memim.com
This is Prof Heiko Braak.
Many years ago, he sat down and examined hundreds of postmortem brains from people with Parkinson’s.
He had collected brains from people who passed away at different stages of the condition, and was looking for any kind of pattern that might explain where and how the disease starts. His research led to what is referred to as the “Braak staging” model of Parkinson’s – a six step explanation of how the condition spreads up from the brain stem (the top of the spinal cord) and into the rest of the brain (Click here and here to read more about this).
 The Braak stages of PD. Source: Nature
The Braak stages of PD. Source: Nature
Braak found that certain populations of cells in the brain were more vulnerable to Parkinson’s than others, such as the dopamine neurons in a region called the substantia nigra, the noradrenergic neurons of the locus coeruleus, and the neurons of the dorsal motor nucleus of the vagus (don’t worry about what any of those names actually mean, I’m just trying to sound smart and make you think that I know what I’m taking about).
One feature that all of these populations of neurons all share in common – in addition to vulnerability to Parkinson’s – is the production of pigment called neuromelanin.
What is neuromelanin?
Neuromelanin is the brain-version of a pigment called melanin, which is found in the skin, eyes, and hair. It is the substance that gives skin & eyes their colour. Dark-skinned people have more melanin in their skin than light-skinned people.
In the brain, certain types of cells, such as the dopamine neurons, produce neuromelanin.

Neuromelanin (the brown patches) in dopamine neurons. Source: Schatz
Neuromelanin appears in large quantities in the human brain, in much lesser amounts in some of the non-human primates, and is almost absent from the brain in many lower species (like mice and rats).
And dopamine neurons in the human brain produce so much neuromelanin that you can visualise it with your bare eye. As you can see in the image below, the Parkinsonian brain has less dark pigmented cells (in the substantia nigra region of the midbrain). As dopamine neurons – which are affected in Parkinson’s – are lost, so too is the dark pigment of neuromelanin.
 The dark pigmented dopamine neurons in the substantia nigra are reduced in the Parkinsonian brain (right). Source:Memorangapp
The dark pigmented dopamine neurons in the substantia nigra are reduced in the Parkinsonian brain (right). Source:Memorangapp
Interresting. How is neuromelanin made?
The mechanism of neuromelanin synthesis are poorly understood, but it is generally agreed that melanin can be made via two methods:
- By the oxidation of dopamine
- By an enzymatic reaction
Sorry I asked. What does any of that actually mean?
So, melanin can firstly be made by the oxidation of dopamine.
What is oxidation?
Oxidation is the loss of electrons from a molecule, which in turn destabilises that particular molecule. Think of iron rusting. Rust is the oxidation of iron – in the presence of oxygen and water, iron molecules will lose electrons over time. Given enough time, this results in the complete break down of objects made of iron.

Rusting iron. Source: Thoughtco
The exact same thing happens in biology. Molecules in your body are constantly going through a similar process of oxidation – losing electrons and becoming unstable.
And this also applies to the protein dopamine.
Once dopamine is made, it is proned to oxidation, and when it is oxidized, dopamine is converted into aminochrome, which undergoes further changes before forming the dark pigment neuromelanin (Click here for a review of this process).
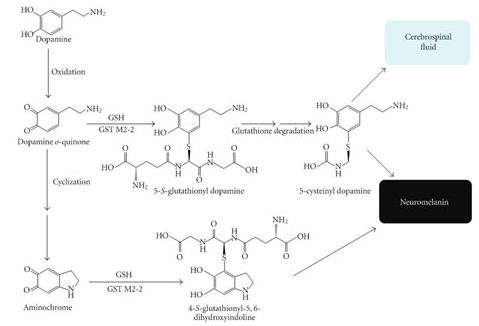 The production of neuromelanin from dopamine. Source: Gale
The production of neuromelanin from dopamine. Source: Gale
Ok, and the second way of making melanin? The enzymatic pathway?
Yes, there is a second way by which melanin can be generated and this is the method that is used outside the brain, in your skin and hair for example. This process involves the oxidation of Levodopa to the melanin precursor DOPAquinone. An enzyme called tyrosinase is responsible for this process.
Is tyrosinase present in the brain?
It has been reported that tyrosinase is present in the human brain:
 Title: Tyrosinase-like activity in normal human substantia nigra.
Title: Tyrosinase-like activity in normal human substantia nigra.
Authors: Miranda, M., Botti, D., Bonfigli, A., Ventura, T. & Arcadi, A.
Journal: Gen. Pharmacol. 15, 541–544 (1984).
PMID: 6441736
In this study, tyrosinase-like activity was found in the substantia nigra of healthy postmortem brain tissue. And this result was replicated by an independent research group (Click here to read more about this).
Interesting. So if tyrosinase is present in the substantia nigra region – which is affected by Parkinson’s – could tyrosinase be involved with Parkinson’s?
That is an interesting question.
One that has just recently been addressed:
 Title: Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis
Title: Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis
Authors: Carballo-Carbajal I, Laguna A, Romero-Giménez J, Cuadros T, Bové J, Martinez-Vicente M, Parent A, Gonzalez-Sepulveda M, Peñuelas N, Torra A, Rodríguez-Galván B, Ballabio A, Hasegawa T, Bortolozzi A, Gelpi E, Vila M.
Journal: Nature Communications 2019 Mar 7;10(1):973.
PMID: 30846695 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers wanted to explore the consequences of inducing high levels of tyrosinase in the dopamine neurons of rats. They achieved this using a carefully engineered virus, in which the disease-causing components of the virus had been removed and the instructions for making tyrosinase were inserted. This made the virus a very effective biological delivery system of tyrosinase. Once the virus infected cells, those cells would start producing high levels of tyrosinase.
Remarkably, 2 months after delivering the virus to the dopamine neurons on one side of the rodent brain, the researchers could clearly visualise dark pigmentation in the dopamine neurons on the injected side of the brain (see the circled area in panel C of the image below). It was only present in the dopamine-producing neurons, and it looked very similar to the neuromelanin found in the dopamine neurons in the human brain.
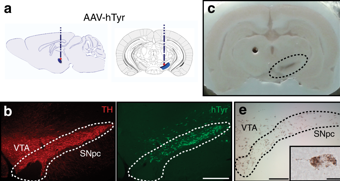 Source: Nature
Source: Nature
By 2 months post virus delivery, the levels of neuromelanin in the rat dopamine neurons reached levels equivalent to those found in the substantia nigra of an elderly human (~80 years old), according to postmortem analysis.
But the cells kept producing neuromelanin, and by 4 months, the levels of neuromelanin were equivalent to those found in post-mortem dopamine neurons from people with Parkinson’s. And it was from this moment that the investigators observed something very interesting: a progressive, age-dependent loss of the neuromelanin producing dopamine neurons!
In the bar graph below, the black bars represent the number of dopamine (TH-positive) neurons on the injected side of the rodent brain. Note the gradual reduction over time.
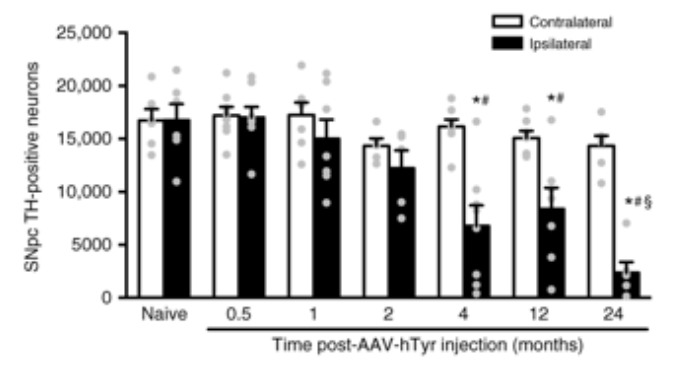 Source: Nature
Source: Nature
And this loss of dopamine neurons was accompanied by impairments to motor behaviour in various tests, AND the accumulation of extracellular neuromelanin (that is, neuromelanin outside of cells – as cells died, they released their neuromelanin into the surrounding environment of the brain. And this last feature was associated with activation of microglia.
What does that mean? Activation of microglia?
Your brain has many different types of cells. Everyone is aware of neurons – the prima donna cells involved with passing messages around the brain – but there are other equally important support/helper cells. Microglia are one of these helper cells. They act as the resident immune cells. When infection or damage occurs, the microglia become ‘activated’ and start cleaning up the area.
 Different types of cells in the brain. Source: Dreamstime
Different types of cells in the brain. Source: Dreamstime
Microglia are constantly monitoring for trouble, and when they detect a problem they will literally be the judge and executioner in deciding if a sick or damaged cell lives or dies. The microglia will change shape as it becomes activated and it will start to release messenger proteins instructing the surrounding cells what to do. Microglia will also alert the immune system, informing it of any problem.
In this current study, the researchers found that microglia were activated when the dying dopamine neurons started releasing neuromelanin, and this has been shown before (Click here to read more about this).
Interesting. Did the researchers find anything else?
Yes they did.
And this is where the story takes an interesting twist.
You see, in addition to neurodegeneration, the researchers also observed Lewy body-like structures.
What are Lewy bodies?
Lewy bodies are dense circular clusters of protein that are characteristically found in specific regions of the brain in people with Parkinson’s (Click here for more on Lewy bodies).
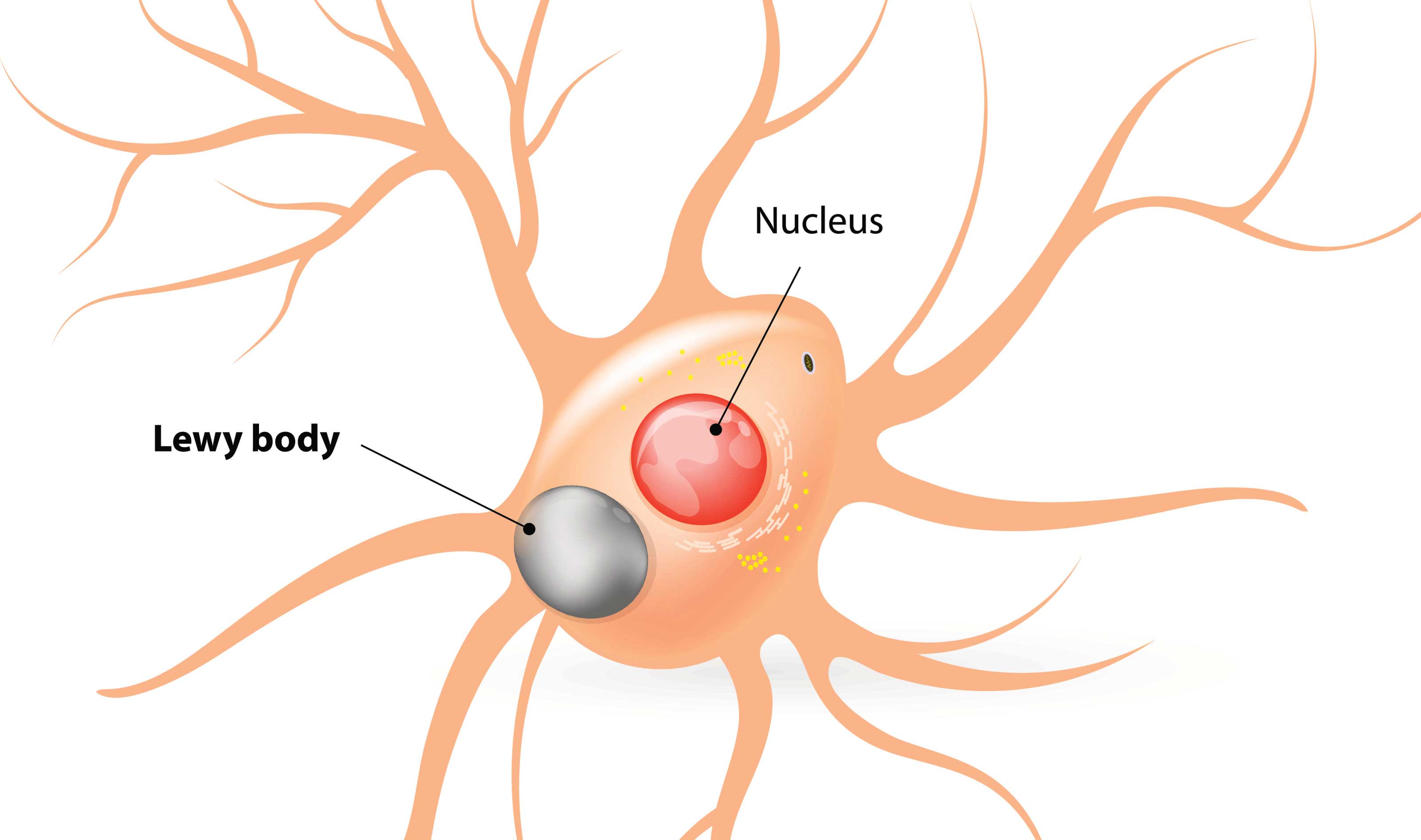 A cartoon of a neuron, with the Lewy body indicated within the cell body. Source: Alzheimer’s news
A cartoon of a neuron, with the Lewy body indicated within the cell body. Source: Alzheimer’s news
The clustered (or aggregated) protein, however, is not limited to just the Lewy bodies. In the affected areas of the Parkinsonian brain, aggregated protein can be seen in the branches (or neurites; see black arrow in the image below) of cells. In the image below, the aggregated protein has been stained brown on a section of brain from a person with Parkinson’s.
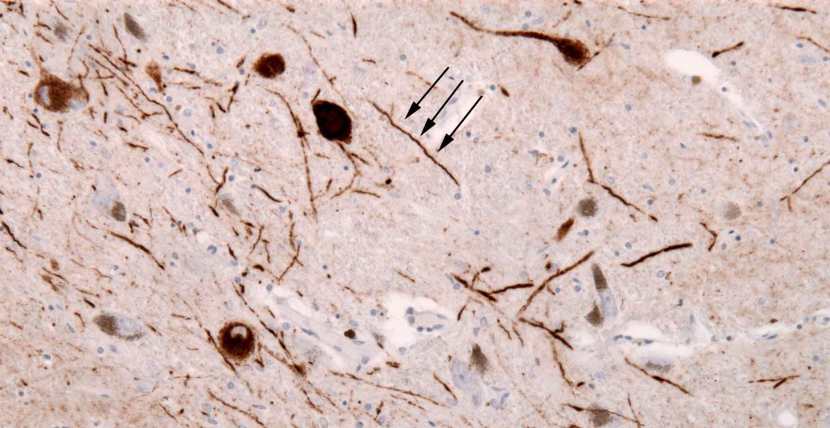
Examples of Lewy neurites (stained in brown; indicated by arrows) from a human brain. Source: Wikimedia
So they researchers found Lewy bodies in the neuromelanin-producing rat dopamine neurons?
Yes. And the Lewy body-like structures could be labelled with many of the appropriate markers of a Lewy body (such as alpha synuclein, Ubiquitin, and p62).
 A rodent dopamine neuron with a Lewy body-like structure, sitting in neuromelanin. Source: Nature
A rodent dopamine neuron with a Lewy body-like structure, sitting in neuromelanin. Source: Nature
But here are the important details regarding these Lewy body-like structures:
- They were completely restricted to neuromelanin-producing dopamine neurons
- Their number peaked at 2 month post virus delivery
- The Lewy body-like structures were substantially reduced by 4 months post virus delivery,…once the neurodegeneration began
Que?
The researchers found that the Lewy body-like structures were most present at 2 months after the delivery of the tyrosinase producing virus. This was before the neurodegeneration actually started.
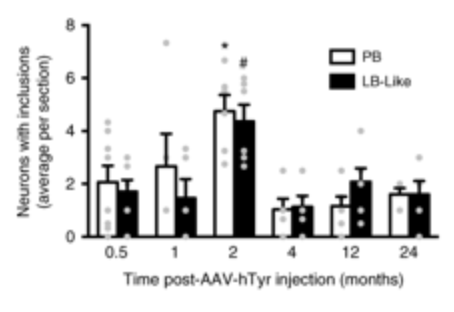 Source: Nature
Source: Nature
In addition, the neurons containing Lewy body-like structures were the ones that preferentially degenerated in these animals.
And this actually agrees with what Prof Heiko Braak originally found in the postmortem brains of people who passed away with Parkinson’s: the number of Lewy bodies in the dopamine neurons of Parkinsonian brains at advanced stages of the condition were much lower than those observed in the earlier stages of PD (Click here to read more about this).
So Lewy bodies are associated with cell death?
I’m not sure.
These results fly in the face of results from other research groups which found the opposite result:
 Title: Contribution of somal Lewy bodies to neuronal death
Title: Contribution of somal Lewy bodies to neuronal death
Authors: Tompkins MM, Hill WD
Journal: Brain Res. 1997 Nov 14;775(1-2):24-9
PMID: 9439824
In this study, the researchers wanted to determine if signs of cell death were more common in dopamine neurons that contained Lewy bodies than dopamine neurons without Lewy bodies. They had previously perfected a staining protocol that allowed them to identify cells in the early stages of apoptosis (or programmed cell death), and they now wanted to use that protocol to assess the role of Lewy bodies.
They stained sections of brain from four postmortem cases (2 cases of Alzheimer’s/Parkinson’s and 2 cases of Dementia with Lewy bodies). Over 1200 neurons were assessed per brain, and what the investigators found was that the total number of neurons that were displaying signs of apoptosis was much greater in the dopamine neurons without Lewy bodies.
That is to say, the majority of dopamine neurons undergoing apoptotic cell death did not appear to contain Lewy bodies (in fact, one of the cases had NO Lewy body-containing dopamine neurons undergoing apoptotic cell death – Lewy bodies were present in the specimen, but there was no sign of cell death in those Lewy bodies-containing cells). This finding led the researchers to conclude that some dopamine cells may be dying before Lewy bodies have a chance to form, which would suggest that the presence of a Lewy bodies does not predispose a neuron to cell death.
And this result was replicated by another independent research group:
 Title: Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease.
Title: Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease.
Authors: Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, Duda JE.
Journal: Neurology. 2012 Dec 11;79(24):2307-14.
PMID: 23152586 (This article is OPEN ACCESS if you would like to read it)
The researchers who conducted this study examined the extent of dopamine neuron dysfunction and degeneration among postmortem sections of brain from 17 healthy controls, 33 with incidental Lewy body disease, and 13 cases of Parkinson’s (with a mean disease duration of 8.3 years). While the density of dopamine neurons (as measured by their total number) was observed to decrease as the Lewy body burden became more severe, a significantly high percentage of dopamine cells were found to be dysfunctional or dying without any Lewy bodies present inside those cells. These results suggest that significant neurodegeneration and cellular dysfunction precede the appearance of Lewy bodies in dopamine neurons,… which basically challenges the idea that Lewy bodies are playing a pathogenic role of Parkinson’s.
And this phenomenon of cell death occurring before protein aggregation does not appear to be specific to Parkinson’s – similar results have been observed in Huntington’s disease (Click here to read more about this).
So, it is fair to say that we are not sure about the role of Lewy bodies. The results of this new study is an interesting finding though.
That’s interesting. So what did they conclude from the study?
Hang on a second, they haven’t finished yet.
The interesting twist in this tale is only just beginning to twist.
One of the proteins that aggregates in Parkinson’s (and forms parts of the Lewy body we discussed about) is alpha synuclein. We have spoke at length about alpha synuclein on this website (Click here to read a recent SoPD post), as it is believed to be one of the chief villans in Parkinson’s.
 Alpha synuclein protein. Source: Wikipedia
Alpha synuclein protein. Source: Wikipedia
And given it’s role of “public enemy number one” in Parkinson’s, the researchers conducting the neuromelanin study decided to test whether it was having a role in the formation of Lewy body-like structures in the neuromelanin-producing dopamine neurons.
They did this by using rats that have been genetically engineered not to produce any alpha synuclein protein. These rats are happy and normal enough, and the absense of alpha synuclein does not appear to cause any issues. So the investigators decided to inject these rats with the ‘tyrosinase-producing virus’ and watch to see what happened.
They may have been expecting to see the absence of Lewy body-like structures and no neurodegeneration, but…
The Lewy body-like structures were still present.
Note the p62 (red) blobs on the right hand side of the image below, which do not have any alpha synuclein (green) colouration. ‘aSynKO’ refers to alpha synuclein knock-out – meaning that alpha synuclein has been removed from the DNA.
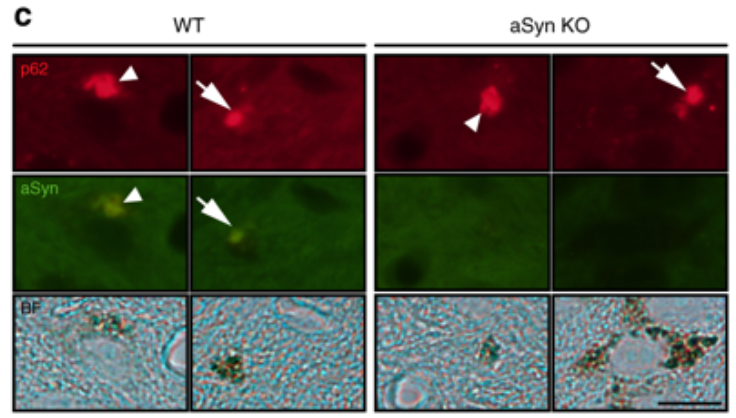 Source: Nature
Source: Nature
IN ADDITION, the absence of alpha synuclein protein had no impact on the neurodegeneration of the dopamine neurons – the same number of dopamine (TH-positive) cells died in both normal (‘wild-type’ or WT) rats as the alpha synuclein knock-out (aSyn KO) rats.
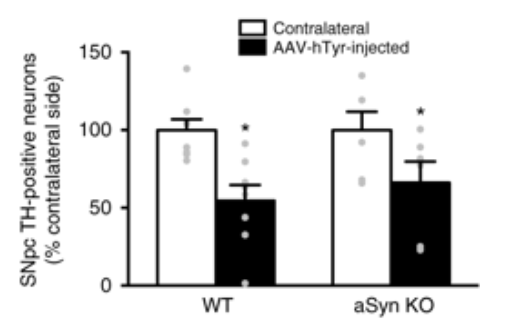 Source: Nature
Source: Nature
This finding suggested that alpha synuclein was not contributing to the neuromelanin-linked cell death in these animals.
Ooohh. Interesting. So summing up?
No, not yet. There is still more!
Given the appearance of Lewy body-like structures in the neuromelanin-producing cells, the researchers wondered whether the recycling/waste disposal system of the cell had been disrupted. Was the build up of clustered protein the result of a failure of the cell to remove old, used protein? Had this vital process become inhibited by the build up of neuromelanin in some way?
Their analysis suggested that it was.
The continuous buildup of neuromelanin ultimately exhausted the waste disposal system (the autophagic capacity) of the dopamine neurons, resulting in a general failure of proteostasis – the maintanence of a correct balance of proteins in a cell – and subsequently the degeneration of neuromelanin-laden neurons.
So if we reduce neuromelanin levels do you think we could slow do this failure and loss of cells?
This is exactly what the researchers asked.
They tested this idea by introducing high levels of a protein called transcription factor EB (or TFEB) in neuromelanin-producing dopamine neurons. TFEB is a master regulator of the recycling/waste disposal system (aka autophagy) – click here for a review of TFEB.
By introducing high levels of TFEB in neuromelanin-producing dopamine neurons, the researchers found that TFEB decreased levels of neuromelanin, markedly reduced the formation of Lewy body-like structures, rescued the loss of dopamine neurons, and improved the performance of the rats in behavioural tests of motor ability.
In the bar graph below, note that raising TFEB levels in rodents injected with the tyrosinase virus (hTyr) results in no significant difference in the number of dopamine (TH-positive) neurons between the injected and control side of the brain (far right column).
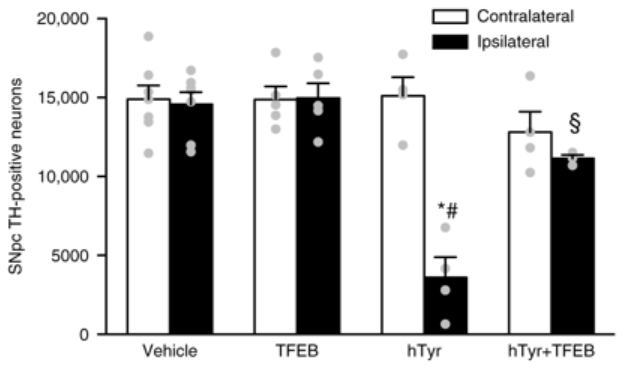 Source: Nature
Source: Nature
Wow! Where can I get me some of that TFEB stuff?
As far as I am aware there are no clinically available activators of TFEB (and I’d be happy to be corrected on this). There have been screening studies, evaluating large libraries of compounds (Click here to read one example).
But of particular interest to our discussion here is this report:
 Title: Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons
Title: Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons
Authors: Magalhaes J, Gegg ME, Migdalska-Richards A, Schapira AH.
Journal: Sci Rep. 2018 Jan 23;8(1):1385.
PMID: 29362387 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers (who are also behind the RAPSODI study we discussed in a previous post – click here to read that post) treated mouse cortical neurons in cell culture with a respiratory medication called Ambroxol and they noted a significant increase in TFEB being activated and shifting to the nucleus of the cells (where it could activate additional waste disposal pathways).
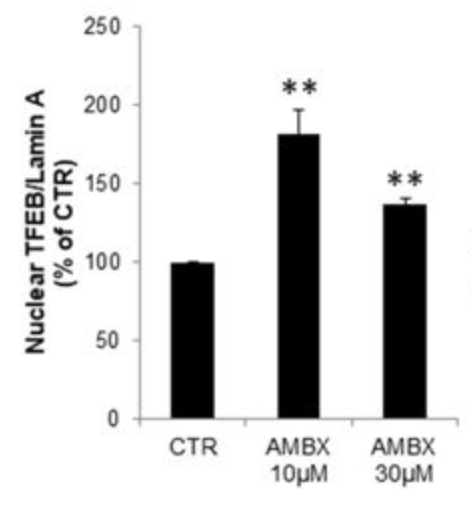 Increase in TFEB in the nucleus after Ambroxol (AMBX) treatment. Source: Nature
Increase in TFEB in the nucleus after Ambroxol (AMBX) treatment. Source: Nature
What is Ambroxol?
Ambroxol is a commonly used treatment for respiratory diseases (the respiratory system being the lungs and related components required for breathing). Ambroxol promotes the clearance of mucus and eases coughing. It also has anti-inflammatory properties, reducing redness in a sore throat. It is the active ingredient of products like Mucosolvan, Mucobrox, and Mucol.

Ambroxol. Source: Skinflint
Researchers have previously reported beneficial effects of ambroxol treatment in models of Parkinson’s, which ultimately lead to a clinical trial.
We are currently awaiting the results of that clinical trial, which is named AiM-PD – Ambroxol in Disease Modification in Parkinson Disease). It was is a phase IIA prospective, single-centre, open label clinical trial to evaluate the safety, tolerability and pharmacodynamic effects of Ambroxol in Parkinson’s (Click here to read more about this trial).
This trial, which is funded by the Cure Parkinson’s Trust and the Van Andel Research Institute (USA), has been conducted at the Royal Free Hospital in London (UK). The study has involved 20 people with Parkinson’s self-administering Ambroxol (in 60 mg per tablet) over a 6 month time frame. The participants were given 5 escalating doses of the drug for the first few weeks of the study (from 60 mg three times per day, gradually building up to 420 mg three times a day after the first month of the study). It will be interesting to see the results of this study later this year.
For those interested in reading more about it, click here for an interesting OPEN ACCESS review of waste disposal enhancing agentsin the context of Parkinson’s.
Is this the first time anyone has ever investigated tyrosinase in the context of Parkinson’s?
No.
Another independent research group published this study more than 10 years ago:
 Title: Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease.
Title: Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease.
Authors: Greggio E, Bergantino E, Carter D, Ahmad R, Costin GE, Hearing VJ, Clarimon J, Singleton A, Eerola J, Hellström O, Tienari PJ, Miller DW, Beilina A, Bubacco L, Cookson MR.
Journal: J Neurochem. 2005 Apr;93(1):246-56.
PMID: 15773923 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers found that tyrosinase is present at very low levels in the postmortem human brain, but they also induced high levels of tyrosinase in cells in culture and found that the treatment made the cells more susceptibility to oxidative stress. They also conducted an anaysis of DNA and found no association between genetic variations in the region of DNA responsible for the production of tyrosinase and increased/decreased risk of Parkinson’s.
But other research groups have reported additional interesting connections to Parkinson’s, such as this report:
 Title: Parkin protects against tyrosinase‐mediated dopamine neurotoxicity by suppressing stress‐activated protein kinase pathways
Title: Parkin protects against tyrosinase‐mediated dopamine neurotoxicity by suppressing stress‐activated protein kinase pathways
Authors: Hasegawa T, Treis A, Patenge N, Fiesel FC, Springer W, Kahle PJ.
Journal: J Neurochem. 2008 Jun;105(5):1700-15.
PMID: 18248610 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers reported that high levels of tyrosinase resulted in increased rates of cell death (apoptosis) in cell cultures. They also reported that the Parkinson’s-associated protein PARKIN could reduce the negative effects of high levels of tyrosinase (Click here to read a previous SoPD post about PARKIN).
So neuromelanin is bad then?
This is the curious part of this story.
Neuromelanin was originally believed to have a protective function.
It was reported that neuromelanin was very good at soaking up inorganic and organic toxins in dopamine neurons (Click here to read more about this). But now this idea appears to be shifting. And as neuromelanin increases with age in the human brain, there may be a threshold at which too much of a good thing, becomes a bad thing.
It will be very interesting to see this research independently replicated and extended by others.
So what does it all mean?
Researchers have published an interesting new study that investigates the role of the pigment neuromelanin in the development of Parkinson’s. Their results suggest that it may have a strong influence.
In a recent SoPD post, we discussed a new evolutionary theory of Parkinson’s. It suggests that some parts of the human brain (such as the region containing the substantia nigra dopamine neurons) have not expanded over evolutionary time as much as other areas of the brain (such as the larger cortical areas of our brain – click here to read that post). This results in an imbalance, because some populations of cells – like the dopamine neurons – have many connections in the newly expanded areas, and this forces them to work harder to do their function. In addition, as we live longer this puts even more pressure on those cells.
 Source: Wiley
Source: Wiley
Now add to that mix the idea that has been discussed in today’s post: as we age certain cells in our brain accumulate neuromelanin. If that neuromelanin has a threshold, beyond which it becomes toxic, this is only going to add further pressure on an already stretched system… and hey-presto, Parkinson’s.
I find this idea quite appealing from the standpoint of late-onset (after 50 years of age) Parkinson’s.
But, while it may explain the loss of certain populations of cells in Parkinson’s which contain high levels of neuromelanin (such as the dopamine neurons in the substantia nigra, the noradrenergic neurons of the locus coeruleus, and the neurons of the dorsal motor nucleus of the vagus – all of these regions are affected by Parkinson’s), it does not explain the loss of other populations of cells in the brain that do not contain neuromelanin (such as the serotonergic neurons of the raphe nucleus and the cholinergic neurons in the nucleus basalis of Meynert – which are also are affected by Parkinson’s). Thus, while I am intrigued by this new research, I am containing my enthusiasm until I see independent replication and extension of the work.
If the results are reproduced, however, it could provide nice support for experimental treatments like Ambroxol which are targetting the recycling/waste disposal system of the cell. If boosting the clearance of rubbish from a cell can help make the cell function better and reduce the chances of the cell dying, this could represent a method of slowing down/stopping the progression of Parkinson’s.
While I don’t want to raise expectations regarding the Ambroxol clinical trial results, from an academic stand-point it will be interesting to see what they suggest.
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from thefourthangelsbowl

{kind=link}
thanks for posting on saturday – now go enjoy the rest of your weekend : )
LikeLiked by 2 people
Thanks Diana – will do! I hope you have a nice weekend too
LikeLiked by 1 person
BRILLIANT! THANK YOU SO MUCH! WE LOVE YOU SIMON!!!
LikeLike
You are very welcome iscatmen.
Kind regards,
Simon
LikeLiked by 1 person
I need to recharge my Lewy body structures LOL
LikeLike
I have to admit I’m a little confused about what this article is saying regarding the role of alpha synuclein (as opposed to neuromelanin) in the PD disease process. Are we saying that Lewy bodies are just a *sign* of the malfunction of autophagy, but that the real impact of that impaired autophagy on the disease process occurs through the buildup of neuromelanin?
I think I read somewhere that *alpha synuclein* sequesters TFEB so that autophagy is impaired. So I guess providing more TFEB via a virus overwhelms that effect, restores autophagy, and then that autophagy reduces the amount of alpha-synuclein, which reduces the suppressive effect that the *AS* has upon autophagy…?
So if not only neuromelanin impairs autophagy, but also alpha synuclein impairs it, then I’m not sure why they would not *both* be significant to the disease process as it occurs *within* neurons.
And outside neurons in the inter-cellular space, since both neuromelanin and AS can activate microglia into the M1 state, they would both seem to be players. And so AS spilled out from a dying neuron that has AS in it could result in the subsequent deaths of bystander neurons that do *not* have any AS, but only have neuromelanin. And vice-versa; neuromelanin-containing neurons, when they die, could spill neuromelanin, activating microglia which then kill some of the AS-containing neurons. Which *maybe* could be why some neurons that do not have any Lewy bodies in them yet are nonetheless dying. I.e., perhaps that does not mean that Lewy bodies are insignificant to the disease process, but only that they are *contributors* to it that need not be present in an individual neuron in order for them to contribute (through autoimmune processes) to the death of that neuron?
We have been focused on addressing the inter-cellular / microglial side of this process for some time now. My partner (who has PD) has been taking curcumin, baicalin and EGCg for years as a means of inhibiting microglial activation from both alpha-synuclein and neuromelanin. We’ve been proceeding on the assumption that damaged and dying dopamine neurons spill out their internal alpha-synuclein and neuromelanin into the inter-cellular space, just as you’ve described above, and that both of these activate receptors on microglia which cause them to transform into M1 (aggressive attack) mode, leading to the deaths of additional “bystander” neurons, which in turn spill their contents, re-starting the auto-immune feedback loop.
The means of neuromelanin triggering microglial activation (at least one such means) occurs through the triggering of the TLR2, CD14 and NOD2 receptors. The following article from 2015 describes the impact of neuromelanin upon those receptors:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4302615/
Curcumin inhibits the TLR2 and NOD2 receptors, and thus we surmised that it might be effective in attenuating the auto-inflammatory feedback loop involving neuromelanin. Here are some articles on that:
https://www.ncbi.nlm.nih.gov/pubmed/24723245
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2593422/
We have a number of other means that we employ to try to slow disease progression, including antioxidants to interfere with the apoptotic caspase cascade triggered by microglia-produced cytokines, and ubiquinol to shore up the mitochondrial energy sources of neurons so that they can defend themselves from attack.
But one thing that we are not yet doing is attempting to stimulate the cleanup of alpha synuclein and neuromelanin using the neurons’ own internal processes. We looked at nilotinib with some interest as a means for stimulating this process through proteasomal enhancement, but have been discouraged by its toxic profile. The Ambroxol trial that your Cure Parkinson’s Trust is helping to study seems like a less nasty way to attain the same goal through enhanced autophagy.
I have always thought that interfering with the PD disease process could ultimately be successful only if a variety of approaches were used; there is no silver bullet. I think we may need to intervene in the disease process internal to neurons, the autoimmune reaction from things dumped into the inter-cellular space, cell energy impairment, alpha synuclein aggregation, and internal cell cleanup processes, all at the same time.
LikeLiked by 1 person
Hi Lou,
Thanks for your interesting and considered response as usual. I have to admit that there were a lot of “Yeah, but…” moments while writing this post and it could have gone off in a dozen different directions. There are quite a few examples of conflicting data, but it is important to remember context – this is an artificial model. I agree with everything you have said, but for the sake of space/time/sanity I tried to keep the storyline here consistent and based around the narrative that the researchers were proposing. This is partly out of respect to them, but also to see what comes out of the wood work in the comments section (and I thank you for your contribution).
Regarding curcumin, although the data is generally positive I have been weary of the poor bioavailability of it. One very good critical viewpoint of it is provided by this review (https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00975) – I would really appreciate your thoughts on this. In every compound I chase, I always seek out the negative/opposing arguments – this review is a good one, providing some balance to the otherwise overwhelming positive data.
And I agree with your inter- plus intra-cellular thinking to treatment of Parkinson’s. It is the best way to approach the problem.
Thanks again,
Simon
LikeLiked by 1 person
[My earlier comment has not yet appeared so I’ll incorporate it with some revisions into this present comment, as the additions will not make sense without that earlier text.]
My very incomplete understanding is that curcumin’s bioavailability issues stem from it being tagged immediately for elimination by the liver through a process called glucuronidation. The form of curcumin that we are using is called Longvida, which was developed by a team at UCLA for the purpose of allowing curcumin to evade this process. It does this by surrounding each tiny nanoparticle of curcumin with a microsopic shell of lipid spheres, which wards off the tagging long enough to get the curcumin into the brain, where it gets trapped in the fatty tissue there, accumulating to even higher levels than can be detected in plasma.
For comparison, one study shows that for a 650mg dose of regular 95% curcumin, peak plasma is undetectable (less than 1 ng/ml) while the same dose of Longvida has a peak plasma level of 22.43 ng/mL:
https://www.researchgate.net/publication/41110422_Safety_and_Pharmacokinetics_of_a_Solid_Lipid_Curcumin_Particle_Formulation_in_Osteosarcoma_Patients_and_Healthy_Volunteers
Although the article you have linked (“The Essential Medicinal Chemistry of Curcumin”) does not mention Longvida curcumin by name, it does mention “nanoparticle systems,” of which Longvida would appear to be the leading example – but apparently only to disparage them, as well as all other attempts at improving bioavailability.
This seems strange, since the central contention of the article is that curcumin, while having many interesting effects in vitro, cannot remain in the body long enough to have any benefits in vivo.
I would have to ask why the authors seem so prepared to write off curcumin for its bioavailability issues, when they acknowledge that it shows so many interesting activities during in vitro testing. Why would they not instead look toward *solving* the bioavailability issues with curcumin? And why did they not *explore* the attempts *already* made to address bioavailability, such as the development of the encapsulation process used to make Longvida curcumin?
Instead, the authors are increasingly dismissive of all such attempts, At one point, they state “[w]e note that lipid dispersions and nanoparticle systems have been developed for 1, with modest improvement in the absorption and bioavailability of the compound.(82)” And that’s all they say there!
First of all, a greater than 22 times improvement in blood plasma levels (perhaps *much* greater, depending on how far below the detection limit plasma levels of regular curcumin were in the linked study above) hardly seems to be a “modest” improvement. And, why do they seem to have so little interest in these efforts?
Later in the article, they note that “[s]ome of the formulations investigated to improve the oral bioavailability of 1 also hope to slow down the observed high rate of metabolism upon absorption. Unfortunately, it appears that once 1 is released in vivo, it has a high potential for modification by both first and second phase metabolism.” Again, very dismissive of these efforts, and no references are given for this contention, nor do they indicate what relationship a “high potential for modification” might have upon the more salient issue of *efficacy*.
Later, we have “Fundamental medicinal chemistry principles, and available ADMET evidence, incline us to hypothesize that the observed high tolerance in humans and low rate of adverse events is likely due to its poor absorption and low bioavailability.” This is a gratuitous “blah blah inclines us to guess the most insulting possible reasons for generally positive traits” kind of comment, which I believe shows naked bias.
Later, more of the same: “Delivery systems such as lipid vesicles, nanoparticles, and nanofibers might be able to boost the bioavailability of 1, but this could also conceivably narrow its therapeutic window and lead to off-target toxicity by aforementioned processes.” Yes, they “could conceivably” do unspecified bad things, and again there is no reference or example illustrating that this possibility is at all likely to occur. Does this sound like an impartial review?
At various points, curcumin is criticized from the perspective of curcumin’s usefulness as a “lead compound” for the development of new drugs, rather than focusing on the usefulness of curcumin itself. The authors criticize curcumin as a “poor lead compound” in a number of places throughout the article. They apparently dislike the fact that curcumin has a number of different modes of action, preferring substances having a more singular and isolable effect. But this multiplicity of effects is actually very useful when you are treating an illness that involves a number of related processes, if the effects happen to synergistically modulate a number of those processes. So the interests of the review’s authors appear to be different from those of the typical PD patient; if a race car driver tells me that my reliable sedan is not a good car for racing, I don’t really care, because the car is serving me very well on my more ordinary commutes about town.
As already mentioned, we are using curcumin as one part of a multi-pronged attempt to slow down PD progression. I think that the in vitro results become relevant if you believe, as I do, that nanoparticle encapsulation has effectively addressed the bioavailability problem. And there are quite a number of in vitro studies showing that curcumin does useful things like inhibiting the aggregation of alpha synuclein.
Also, there *are* *some* in vivo results (albeit with rodents) which also suggest that curcumin can play a role within that overall treatment approach.
Here, for example, is a rodent study that shows that curcumin inhibits TLR 2 and 4 receptors on microglia:
https://www.ncbi.nlm.nih.gov/pubmed/24723245
From the above article:
“Curcumin significantly reduced neurological deficit scores, cerebral infarct size, neuronal damage, cerebral water content, and MPO activity. It also inhibited the expression of TLR2/4 and decreased the expression and activity of NF-κB p65 in rat brain. In addition, curcumin attenuated the release of TNF-α and IL-1β in blood. Our results suggest that curcumin reduces inflammatory reaction and brain damage in a rat model of permanent focal cerebral ischemia.”
And, here is a rodent study that shows that a component of curcumin induces neural stem cell proliferation:
https://stemcellres.biomedcentral.com/articles/10.1186/scrt500
I would note that none of the (rather odd and sparse selection of) studies cited in the article as showing curcumin’s lack of efficacy, used a nanoparticle formulation; they apparently all used regular curcumin extract. And the study authors pretty much said that they were not surprised at the lack of efficacy because of curcumin’s bioavailability issues.
So, the main issue seems to be whether curcumin can be made bioavailable, and the authors of the study have pretty much avoided any substantive exploration or discussion of that very issue.
Given the above, I have to say that I do not find this study to be impartial or objective.
Now, there’s a lot of detail in this article that I’m certain I have not absorbed. And if some of what I have missed should give me pause in concluding what I have in the above remarks, I hope that you will point it out to me.
LikeLiked by 1 person
This response to the linked article, titled “Curcumin May Defy Medicinal Chemists,” may be of interest:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5346972/
And here is the reply of the authors, titled “Curcumin May (Not) Defy Science,” to the above response:
https://pubs.acs.org/doi/full/10.1021/acsmedchemlett.7b00139
Interestingly, in this reply, they discount a study that shows efficacy because it uses a highly bioavailable form of curcumin that (no doubt because of the techniques used to attain that bioavailability) is only 30 percent curcumin:
“The ‘curcumin’ and COPD trial published by Funamoto et al. is a double-blinded, placebo-controlled study.(4) However, it employs Theracurmin, a ‘highly-bioavailable’ form of turmeric extract, which contains approximately 30% curcumin and therefore cannot be simplified as being curcumin. This, again, severely decreases strength of an argument for curcumin as the therapeutic agent and is consistent with the TEMCC content.”
Now, maybe from the standpoint of drug development this makes some kind of sense. But from the standpoint of patients evaluating the usefulness of curcumin in some bioavailable formulation, it does not make any sense at all. If these folks cannot at least make an attempt to distinguish the effects of a delivery system from the drug itself, concluding when appropriate that the delivery system has a neutral effect, then it would seem that they’re after something very different from what my partner and I are seeking as a caregiver and patient.
They also say that “Interestingly, by stating that curcumin can only be observed transiently in the plasma of animals, the authors of the letter confirm that curcumin itself can be excluded as the pharmacologically active agent.” However, I have read that brief repeated exposures to curcumin in plasma can build up in the brain to levels quite a bit higher than those present in the plasma. This study, for example, shows (see TABLE 1) that levels of curcumin in plasma when mice were fed curcumin by gavage were not detectable, but levels in the brain were nonetheless detected:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2527621/
LikeLike
I just want to add that an article like the one linked above makes me appreciate this site all the more, because I feel that the linked article uses technical information to exhaust and overpower its readers in the service of the views held by its authors, while the purpose of this site is to clarify difficult technical material, thus empowering its visitors.
LikeLiked by 1 person
I’m a fan or turmeric/curcumin; I take curcumin supps every day. Usually I take a whole turmeric extract with piperine (Gaia Herbs) in the am and Theracurumin micronized curcumin in the pm, since I like to hedge my bets. The Gaia Herbs product has had a noticeable effect on my mood, while the Thercurmin product has some clinical studies.
That being said, I have not seen any clinical trials or in vivo animal studies that really match the ‘wow’ results of the in vitro studies, except perhaps for this one, where they injected curcumin into the eyes of mice:
https://www.karger.com/Article/FullText/485085
A while back some there were some exciting reports on a curcumin derivative, J147:
https://www.salk.edu/news-release/alzheimers-drug-turns-back-clock-powerhouse-cell/
LikeLike
Thank you so much for this amazing blog. I am caring full time for a father with Lewy Body Dementia (and a mother with Alzheimer’s). So much of the Parkinson’s research is applicable to LBD, so I find reading your blog very helpful. I have cautiously added some of the researched supplements to Dad’s diet, and although it impossible to know for sure if they have had a positive effect, he has had no progression of his motor symptoms for the past 4 years–and has actually improved significantly in his mood and cognitive abilities. His executive function remains compromised, but his apathy and depression have completely disappeared and he is able to enjoy reading and watching sports each day. I did start him on ambroxol (have to import from Germany), about 4 months ago, albeit a lower dose than is being studied (he’s up to 450mg a day.) He does not take any dopamine (or agonists) as it cause it hallucinations when we tried it 4.5 years ago. He does take Rivastigmine 12mg, extended release 5mg melatonin at bedtime and a combo of Mirtazapine 30mg at bedtime and very low does venlafaxine (37.5mg) in the am (an antidepressant called “California Rocket Fuel”. Those doses have been unchanged for the past 4.5 years (when he was diagnosed with LBD.) He is remarkably stable from day to day.
LikeLiked by 1 person
I am also using Bits of Advice from this absolutely brilliant Scientist and Humanist Simon, He is simply life-saving for our parents around the globe. I am using information for treatment of my 82 years old LBD mommy from 2.5 years very very carefully and we have also very stable positive results last 18 months. She is watching TV now and also starts cook in the kitchen and this is unbelievable!. I am giving my mommy (2 years) 2.5 mg/night Melatonin + very low dose Clonarex (Clonazepam) 1/4 of 0.5mg/night for REM phase. She has only 4-5 times 3D hallucinations last 12 months ( before it was 2-3 times/week ) Day time for the movement she is taking 1/2 of 125mg Madopar and 100mg natural Mucuna Prurience (velvet been) and for mitochondria 500mg Niacin (vitamin B3) She is taking Niacin 30 days and then rest 30 days and so on. She is taking regularly natural antioxidants ( red wine Resveratrol) and Curcuma
I am considering to start Ambroxol and stop any Levodopa (Dopamine).
JJ could You please kindly explain to me how exactly you take and evaluate/change the dose of Ambroxol ? last 4 months.
I will be very grateful for your advice,
My email is info@imscat.men
God Bless Simon
LikeLike
Hallucinations and delusional behavior tend to be associated with damage to cholinergic function. You might look into Alpha GPC (aka choline alphoscerate) as a way of raising brain acetylcholine levels, which can help to compensate for such damage.
My father was never delusional, but he developed Mild Cognitive Impairment in his late 80s and benefited from taking 300mg of Alpha GPC 4 times daily. Here’s a review article on its usefulness to demential patients:
Click to access defbd50c793d4a34ea099ff94fe182a45b45.pdf
From that article:
“Controlled clinical trials reviewed in this paper have demonstrated the efficacy of choline alphoscerate in clinical situations associated with cognitive impairment characteristic of mild cognitive impairment (MCI) or even dementia disorders, both of degenerative and vascular origin. The stated therapeutic usefulness of choline alphoscerate in the relief of cognitive symptoms, such as memory and attention impairment, differentiates the drug from cholinergic precursors (lecithin, CDP-choline) used in former clinical trials. The results of uncontrolled trials carried out in the treatment of TIA or stroke suggest that choline alphoscerate might favor functional recovery of patients with acute cerebrovascular event. Although these findings need to be confirmed by further controlled trials, published clinical data collectively suggest a clinical efficacy of this cholinergic precursor in cognitive impairment occurring in the elderly.”
In particular, this paper compares Alpha GPC favorably with acetylcholinesterase inhibitors, some examples of which are rivastigmine and donepezil (Aricept).
“[T]his trial, different from previous studies with choline alphoscerate has used batteries of tests and time of observation comparable with studies assessing the activity of (acetyl) cholinesterase inhibitors. A comparison of ADAS-Cog analysis from this investigation, with the results obtained on the same item in 4 trials with the cholinesterase inhibitor donepezil revealed a more positive trend with the cholinergic precursor choline alphoscerate compared with donepezil (Fig. 3)…”
Just have a look at the chart in Fig. 3; it shows an end point after six months of treatment with three times the benefit (on the Adas-Cog test) for Alpha GPC as for donepezil. Lower scores are better for that test, which measures cognitive impairment.
I use it myself for middle-aged memory impairment, and the results have been significant and beneficial.
LikeLiked by 1 person
Ich wohne in Deutschland und werde Ambroxol demnächst ausprobieren.
LikeLike
we wish you a very best luck, my mommy 82 (LBD) start the trial 4 days ago still 181 to go
LikeLiked by 1 person
I hope I am not being totally ignorant, but I have read that there is a higher incidence of melanoma in people with PD, despite a lower incidence of other cancer types, and that there may be a common cause. I also read that one skin cancer drug may be of use in PD, but has nasty side effects and is very expensive. Melanin and neuromelanin have similar chemistry. So if there is some hope that ambroxol may help clean up mechanisms – has anyone looked at this from the skin cancer side? There may be bugger budgets for cancer research……
A separate question, why is ambroxol not available in the UK, but easily available in mainland Europe in OTC medicines?
LikeLiked by 1 person
Yep. Overproduction of melanin/neuromelanin may well be what explains the higher prevalence of melanoma in Parkinson’s patients. Neuromelanin is a major activator of microglia, and there are several times as many microglia in the substantia nigra as in other areas of the brain. So once neurons start to die, they dump their neuromelanin into the surrounding environment, where they stimulate microglia to convert to M1 “attack” mode, injuring bystander neurons that were not previously damaged, and thus feeding the autoimmune inflammatory process that causes major neuronal casualties in PD.
Since the Miro1 work published at the end of 2019 indicates that something is preventing Miro1 from dissolving when it otherwise would, to allow unhealthy mitochondria to which it is attached to be garbage collected, and since the same dysfunctional behavior is present in skin cells of 94 percent of Parkinson’s patients, this makes me wonder if neuromelanin might not also be playing a role in preventing the Miro1 protein from dissolving when it should.
LikeLike
Just when you were confident that neuromelanin meant protection along comes evidence suggesting otherwise!
The mid brain ‘with and without’ images recalls a study, see following summary, of the nigrosome sub level of the substantia nigra. This also by inference relates to the recent discussion about UPDRS or more fundamentally the critical need for an in vivo biomarker providing early, accurate cost effective diagnosis and progress tracking. .
Title: The ‘Swallow Tail’ Appearance of the Healthy Nigrosome – A New Accurate Test of Parkinson’s A Case- Control and Retrospective Cross-Sectional MRI Study at 3T Stefan T. Schwarz1*, Mohammed Afzal2, Paul S. Morgan3, Nin Bajaj4, Penny A. Gowland5, Dorothee P. Auer1 PLOS one open access; April 7 2014
Given that much of the current diagnoses comprise motor features as the basis of the clinical diagnosis, establishing the clinical diagnosis can be challenging in early or tremor dominant cases. The reported diagnostic error rates are 4-15% in clinical trials and up to 25% in community studies. DaTscan, remains the only licensed PD diagnostic but these types of scans are expensive, involve low dose radiation and are only available in specialised centres..
Translating this technique to 3T – MRI platforms in order to support the diagnosis of PD would be highly desirable, as MRI at 3T is widely available and much less expensive than licensed nuclear medical techniques. It additionally offers the opportunity for combining diagnostic confirmation of nigral degeneration in PD with promising MRI techniques for differentiating other parkinsonian conditions such as PSP-P, MSA-P and vascular parkinsonism
Two raters independently classified subjects into PD and non-PD according to absence or presence of nigrosome-1. . Diagnostic accuracy of the retrospective cohort was: sensitivity 100%, specificity 95%, NPV 1, PPV 0.69 and accuracy 96%
The study demonstrated that standard high resolution susceptibility weighted MRI at 3T allows the detection of the healthy nigrosome-1, and its absence in PD, yielding a high diagnostic accuracy in a clinical population.
The healthy nigrosome-1 can be readily depicted on high-resolution 3T – SWI giving rise to a ‘swallow tail’ appearance of the dorsolateral substantia nigra, and this feature is lost in PD. Visual radiological assessment yielded a high diagnostic accuracy for PD vs. an unselected clinical control population.
Assessing the substantia nigra on SWI for the typical ‘swallow tail’ appearance has potential to become a new and easy applicable 3T MRI diagnostic tool for nigral degeneration in PD.
A Paradox in Parkinson’s research is illustrated right here.
addressing the same area of the brain, we have:
an image based diagnostic process based on the ‘healthy substantia nigra’ providing a significant, accurate diagnosis with the potential to expand into in vivo monitoring and assessment
a number of research study outcomes demanding a closer investigation of proteostasis and neuromelanin, with potential to identify repurposing opportunities
May you live in interesting times!
LikeLiked by 2 people
Of course, since the results of Xinnan Wang et. al. were published at the end of 2019, we now have a likely marker for PD in the form of a test for whether the Miro1 protein attached to the mitochondria of a patient’s extracted skin cells dissolves in response to the CCCP toxin being used to damage those cells. If it fails to dissolve, it’s a strong indicator that the patient either has or will likely develop PD. This has not become generally available but research indicates that it will probably work.
LikeLike