
|
# # # # Over the last decade, a large number of clinical trials involving immunotherapy have been conducted in the field of Alzheimer’s research. The overall success rate of these studies has not been encouraging. Immunotherapy involves artificially boosting the immune system so that it targets of particular pathogen – like a rogue protein in the case of Alzheimer’s – and clears it from the body. Recently, preclinical research has pointed to several possible reasons why this approach may be struggling in the clinical trials, and potential solutions that could be explored. In today’s post, we will review two research reports and consider how this applies to Parkinson’s research. # # # # |
 Immune cells (blue) checking out a suspect cell. Source: Lindau-nobel
Immune cells (blue) checking out a suspect cell. Source: Lindau-nobel
Immunotherapy is a method of boosting the body’s immune system to better fight a particular disease. Think of it as training the immune cells in your body to target a particular protein.
The approach involves utilising the immune system of your body, and artificially altering it to target a particular protein/disease-causing agent that is not usually recognised as a pathogen (a disease causing agent).
It is truly remarkable that we have gone from painting on cave walls to flying helicopters on Mars and therapeutically manipulating our body’s primary defense system.
Immunotherapy is potentially a very powerful method for treating a wide range of medical conditions. To date, the majority of the research on immunotherapies have focused on the field of oncology (‘cancer’). Numerous methods of immunotherapy have been developed for cancer and are currently being tested in the clinic (Click here to read more about immunotherapy for cancer).
 Many approaches to immunotherapy against cancer. Source: Bloomberg
Many approaches to immunotherapy against cancer. Source: Bloomberg
Immunotherapy has also been tested in neurodegenerative conditions, like Alzheimer’s and more recently Parkinson’s. It typically involves researchers carefully designing antibodies that target a rogue protein (like beta amyloid in Alzheimer’s and alpha synuclein in Parkinson’s) which begin to cluster together, and this aggregation of protein is believed to lead to neurotoxicity.
 Source: RND
Source: RND
What are antibodies?
Antibodies are Y-shaped proteins that the immune system naturally and continuously produces to identify anything in the body that is ‘not self’ (that is, not a normally occurring part of you – think of viruses, bacteria, etc).
 A schematic representation of an antibody. Source: Wikipedia
A schematic representation of an antibody. Source: Wikipedia
These antibodies act like red flags for the immune system. They bind to specific targets, alerting the immune system to entities that should not be there (again, think of viruses or bacteria). Specialised immune cells are on the look out for bound antibodies, and they are ruthlessly efficient at removing anything that they are attached to.
Have there been many clinical trials of immunotherapy in neurodegenerative conditions?
There have been for Alzheimer’s, and unfortunately, the success rate of those clinical trials testing antibody-based immunotherapies in Alzheimer’s has been extremely poor. Only one immunotherapy (Aducanumab) has been approved for the treatment of Alzheimer’s (Click here to read more about this). Many other Alzheimer’s immunotherapy approaches have failed to demonstrate any effect on the progression of the condition.
The immunotherapies have been rather good at clearing the targeted rogue proteins from the brain (despite limited access to the CNS due to the blood brain barrier), but this clearance has not been associated with subsequent clinical improvements.
As a result of this track record, many researchers have wiped their hands of immunotherapy as a treatment for neurodegeneration, but other more intrepid researchers have been digging deeper into the situation and trying to determine what could be going wrong.

Source: Genengnews
And those efforts have led to some interesting recent findings.
What have the researchers found?
In April of this year, this report was published:
 Title: Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia.
Title: Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia.
Authors: Trudler D, Nazor KL, Eisele YS, Grabauskas T, Dolatabadi N, Parker J, Sultan A, Zhong Z, Goodwin MS, Levites Y, Golde TE, Kelly JW, Sierks MR, Schork NJ, Karin M, Ambasudhan R, Lipton SA.
Journal: Proc Natl Acad Sci U S A. 2021 Apr 13; 118 (15): e2025847118.
PMID: 33833060
In this study, the researchers grew human dopamine neurons in cell culture. Dopamine neurons are a type of cell population in the brain that is particularly vulnerable in Parkinson’s. By the time a person starts to exhibit the motor symptoms of PD, they have typically lost 50% of their dopamine neurons.
In this study, half of these dopamine neurons being grown in culture were carrying a genetic mutation (called A53T alpha synuclein) which is associated with an increased risk of developing Parkinson’s, while the other cells did not carry this mutation.
The investigators also grew microglia in the same culture dish as the dopamine neurons, and they looked at what impact the A53T alpha synuclein mutation in the neurons had on the behaviour of the microglia.
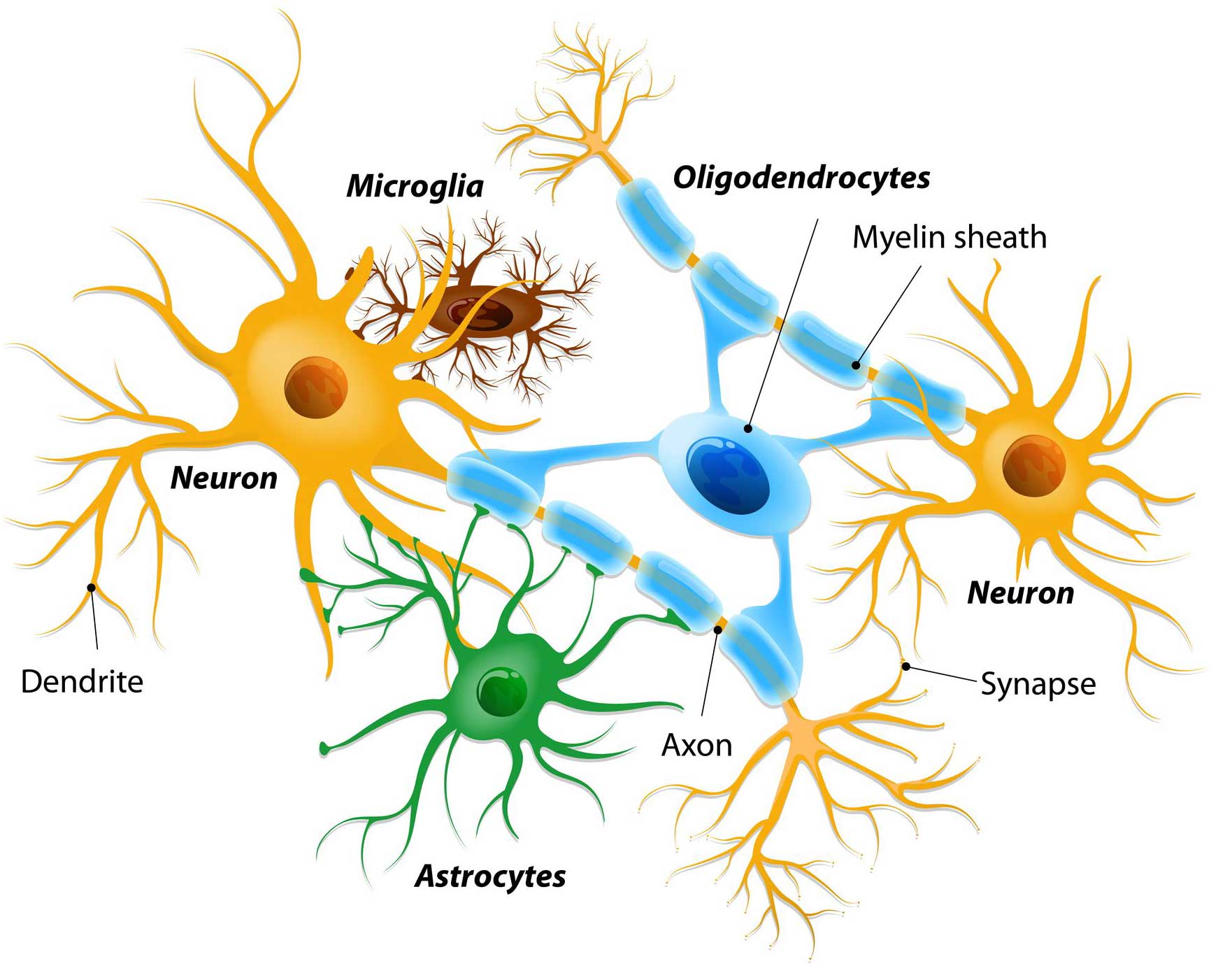
What are microglia?
Microglia are some of the helper cells in the brain, they provide supportive services to neurons. Specifically, they act as the resident immune cells. When infection or damage occurs, the microglia become ‘activated’ and start cleaning up the area.
 Different types of cells in the brain. Source: Dreamstime
Different types of cells in the brain. Source: Dreamstime
When they become activated, microglia do generally do three things:
1. They change shape – microglia usually have outstretched branches when they are in their ‘resting’ state. These branches are constantly monitoring the surrounding environment for any signs of trouble. But when trouble appears, microglia will become activated and retract their branches, giving them a more spherical appearance.
 Source: Diacomp
Source: Diacomp
2. They release cytotoxic proteins – these toxic messenger proteins (called cytokines) encourage a wounded or sick cell to die, helping the microglia to determine which cells are too sick/damage to survive and need to be removed.
 Source: Sigmaaldrich
Source: Sigmaaldrich
3. They start to be very active with regards to phagocytosis.
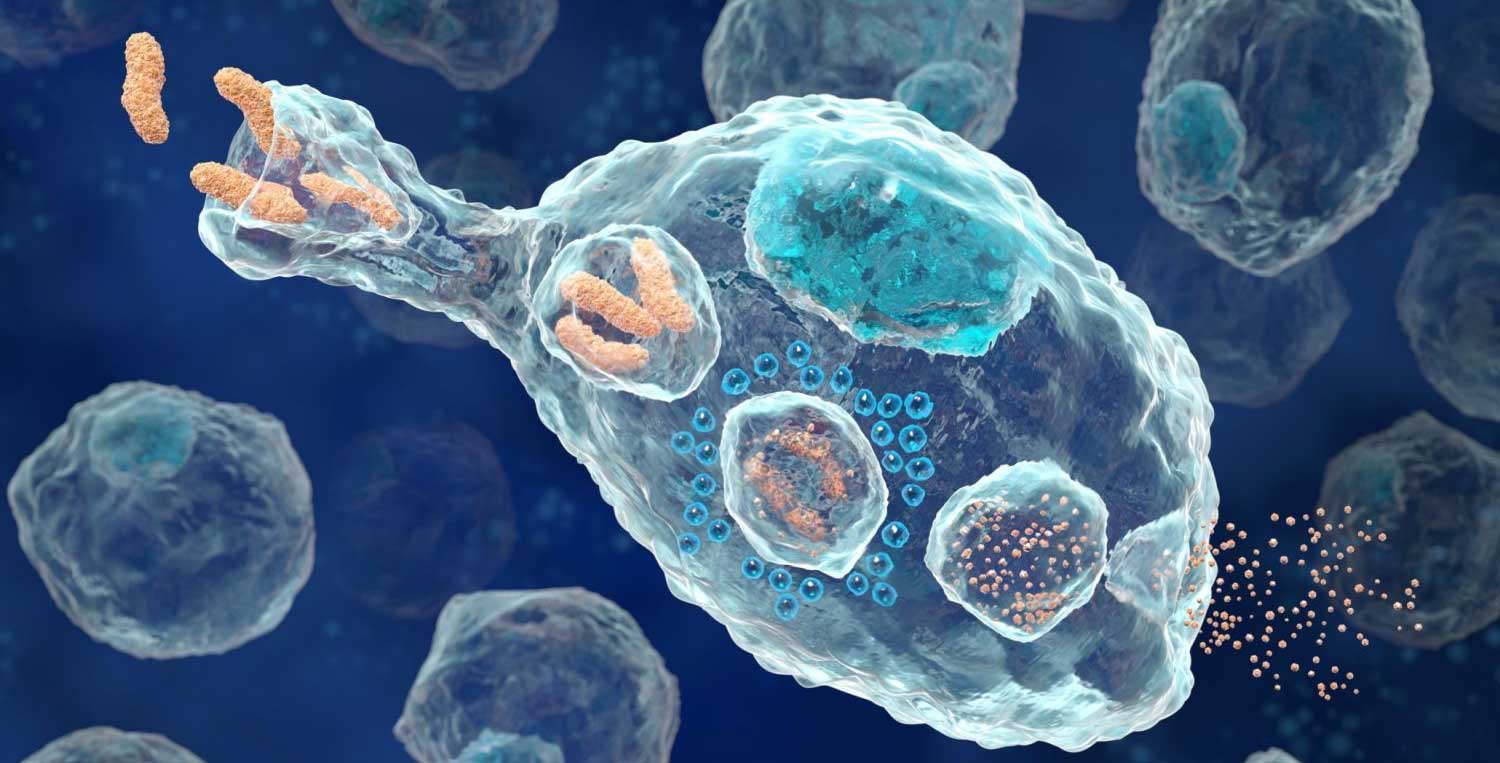
What is phagocytosis?
Phagocytosis comes from Ancient Greek φαγεῖν (phagein) , meaning ‘to eat’, and κύτος, (kytos) , meaning ‘cell’. It is used in biology to refer to the process of engulfing or consuming objects.
 A schematic of a macrophage. Source: Meducator
A schematic of a macrophage. Source: Meducator
Microglial phagocytosis is a process by which dying cells/debris/rubbish can be vacuumed up, broken down and disposed of (Click here to read a good review of microglia-based phagocytosis in the context of Parkinson’s).
By doing this amazing clean up job, microglia are able to help maintain ‘homeostasis’ in the brain.
Got it. So what did the researchers find?
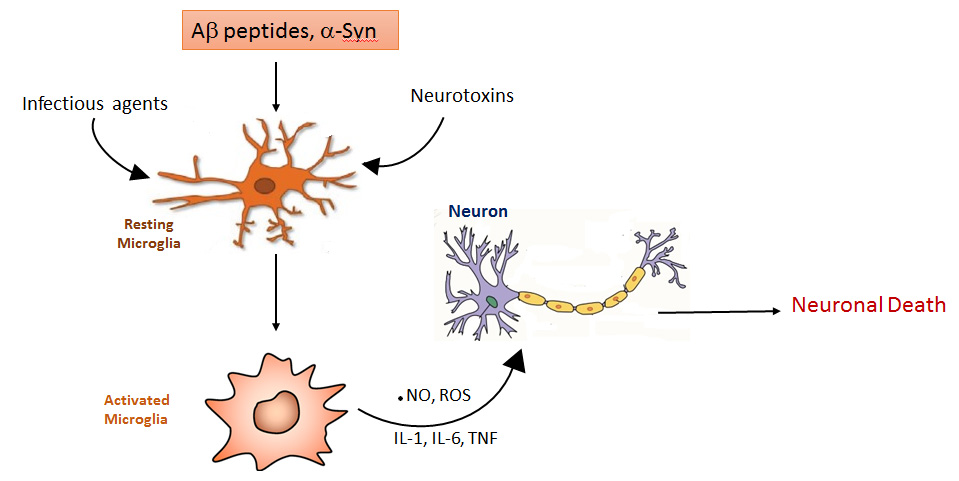
When the investigators grew the microglia cells with neurons that carried the A53T alpha synuclein mutation, they found activation of the NLRP3 inflammasome in the microglia.
What does that mean? What is the NLRP3 inflammasome?
When cells in your body are stressed or sick, they begin to release tiny messenger proteins (these are the cytokines I mentioned above) which inform the rest of your body that something is wrong.
When enough of these messenger proteins are released that the immune system becomes activated, it can cause inflammation.
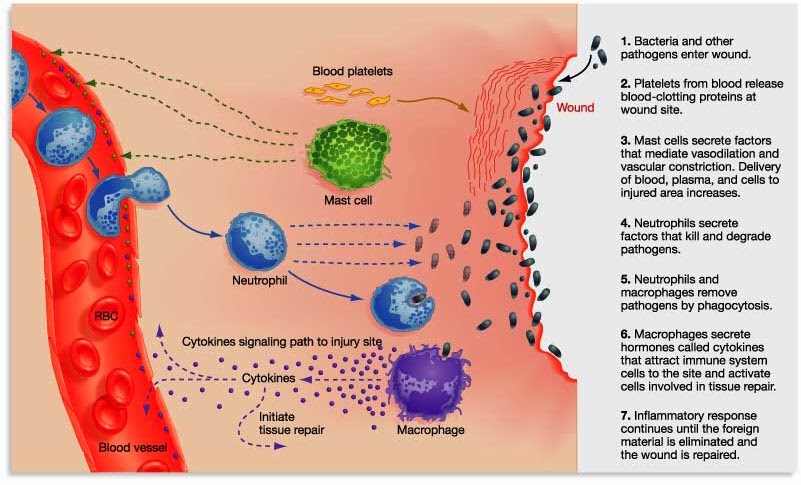
Inflammation is a critical part of the immune system’s response to trouble. It is the body’s way of communicating to the immune system that something is wrong and activating it so that it can help deal with the situation.
By releasing the cytokines, injured/sick cells kick off a process that results in multiple types of immune cells entering the troubled area of the body and undertaking very specific tasks.
 The inflammatory process. Source: Trainingcor
The inflammatory process. Source: Trainingcor
The strength of the immune response depends on the volume of the signal arising from those released cytokines. And there are processes that can amplify the immune response.
One of those processes is called inflammasomes.
What are inflammasomes?
Inflammasomes are multi-protein formations that are present inside of cells in your body. They detect pathogenic agents or stressors that have found their way inside of cells, and once these inflammasomes detect something that should not be there, they activate the release of highly pro-inflammatory cytokines (such as interleukin-1b (IL-1b) and interleukin-1b (IL-18)).
These cytokines are released into the world outside of the cell and amplify the signal to the immune system that something is not quite right.
 Source: Youtube
Source: Youtube
There are difference types of inflammasomes and they vary based on what activates them. For example, the presence of RNA from a particular virus may activate one type of inflammasome, while a certain toxin will cause the assembly of a different inflammasome.
For the purpose of keeping things simple in today’s post, we are going to focus on one of the most well characterised inflammasomes.
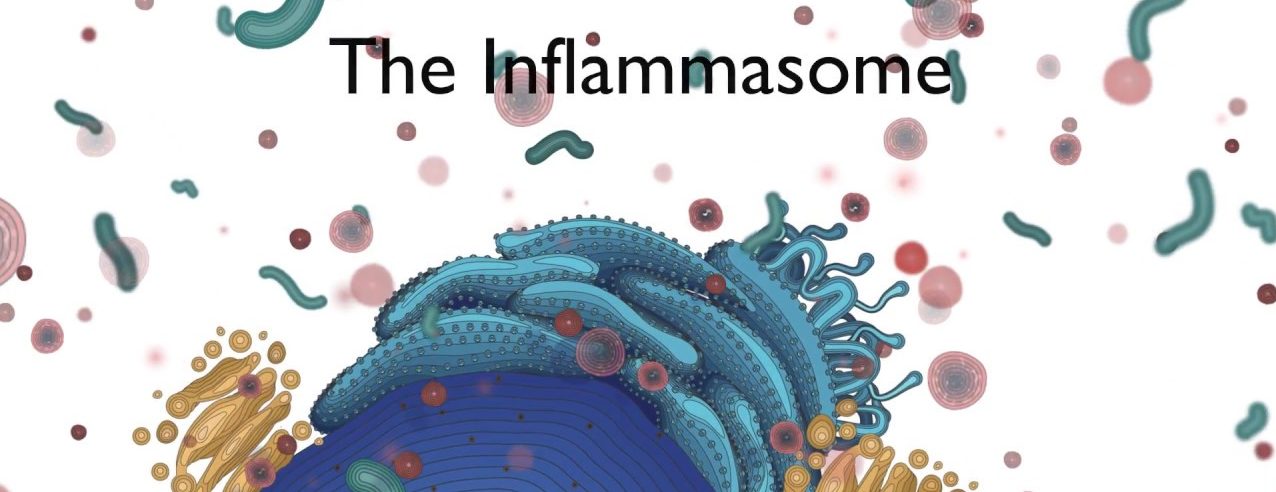
It is called the NLRP3 inflammasome:
 The NLRP3 inflammasome. Source: Twitter
The NLRP3 inflammasome. Source: Twitter
The name of this particular inflammasome is derived from one of the three core components: a protein called NLRP3.
What is NLRP3?
NLRP3 is an abbreviation that is a lot easier to say than “Nucleotide-binding domain, Leucine-Rich-containing family, Pyrin domain-containing-3“.
Ok, so neurons with mutant alpha synuclein activate an inflammation amplification system called the NLRP3 inflammasome. Got it. What did the researchers do with this information?
Firstly, they demonstrated that the clustered (or aggregated) form of this mutant alpha synuclein was activating the NLRP3 inflammasome, by exposing the microglia cells directly to the aggregated alpha synuclein protein. When they did this, the NLRP3 inflammasome became activated and the microglia began releasing pro-inflammatory cytokines.
Next, to demonstrate that the activation of the NLRP3 inflammasome in the alpha synuclein-exposed microglia was resulting in the release of pro-inflammatory cytokines, the researchers administered a drug that inhibits NLRP3 (called MCC950 – we have previously discussed this drug on the SoPD website – click here to read more about this). They found that the drug blocked the activation of the NLRP3 inflammasome and reduced the levels of pro-inflammatory cytokines released.
To further explore the interaction between alpha synuclein and the NLRP3 inflammasome, the investigators next attempted to reduce alpha synuclein levels using an immunotherapy treatment (like we discussed above).
And this is where they discovered something really interesting.
What did they discover?
When the researchers added both aggregated alpha synuclein and the immunotherapy treatment (alpha synuclein-targeting antibodies) to their microglia cells in culture, rather than reducing levels of pro-inflammatory cytokine release, this treatment unexpectedly increased levels of pro-inflammatory cytokines.
That is to say, the immunotherapy treatment appeared to increase inflammation.
Further analysis suggested that the effect was specific to the aggregated form of alpha synuclein being bound to the antibody. The researchers reported that the unaggregated form of alpha synuclein bound to an antibody did not have any effect, nor did the antibody just by itself).
Weird. What did the researchers do next?
Curious to see if this effect was specific to just Parkinson’s-associated alpha synuclein and not to other neurodegeneration-associated aggregated protein, the researchers next exposed their microglia cells in culture to the aggregated form of the Alzheimer’s-associated protein beta amyloid (Click here to read a previous SoPD post about beta amyloid).
The researchers found that aggregated beta amyloid had no impact on the production of inflammatory cytokines, but it did have an effect when given in combination with a beta amyloid-targeting immunotherapy (curiously, there was also a synergistic effect when given in combination with aggregated alpha synuclein – a combo of aggregated beta amyloid & aggregated alpha synuclein).
Considering that these findings could have important implications for ongoing immunotherapy clinical trials in both Parkinson’s and Alzheimer’s, the researchers next wanted to check if this effect could also be observed in animal models of the conditions. So they injected mice with either human microglia cells alone, human microglia cells together with aggregated alpha synuclein, or human microglia cells together with aggregated alpha synuclein and an alpha synuclein-targeting antibody.
 Two weeks later, they found that the human microglia cells injected together with aggregated alpha synuclein resulted in inflammasome activation and neurotoxicity, and this effect was exacerbated by alpha synuclein antibody.
Two weeks later, they found that the human microglia cells injected together with aggregated alpha synuclein resulted in inflammasome activation and neurotoxicity, and this effect was exacerbated by alpha synuclein antibody.
Interestingly, resident mouse microglia in the mouse brain did not exhibit the same effect indicating that the enhanced inflammation in response to aggregated alpha synuclein + antibody complex may be unique to human microglia.
The researchers concluded from their study that negative findings of many immunotherapy clinical trials could in part be explained by a microglial inflammatory response to the antibody+protein complex that forms from the treatment. Further research is obviously required, but if independently replicated, we may need to start looking at the possibility of evaluating some kind of anti-inflammatory approach with future immunotherapy treatments.
|
# RECAP #1: Inflammation is the body’s way of letter the immune system know that something is wrong. This response can be amplified via a mechanism called the inflammasome. Immunotherapy is a technique that could be used to treat medical conditions, like Alzheimer’s or Parkinson’s. It involves manipulating the immune system to target specific pathogens (disease causing entities). New research, however, asks whether there could be an inflammatory response to some immunotherapies. # |
Interesting, so summing up? What does it all mean?
No, not just yet.
There is another recent immunotherapy-based study that also deserved our attention.
This report was also published in April this year:
 Title: Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy
Title: Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy
Authors:
Journal: Nature, 28 April 2021. Online ahead of print.
PMID: 33911285
In this report, the researchers wanted to explore whether a genetically engineered mouse model of Alzheimer’s (called 5xFAD mice) exhibited any lymphatic dysfunction over time with the development of Alzheimer’s-like pathology.
What is lymphatic dysfunction?
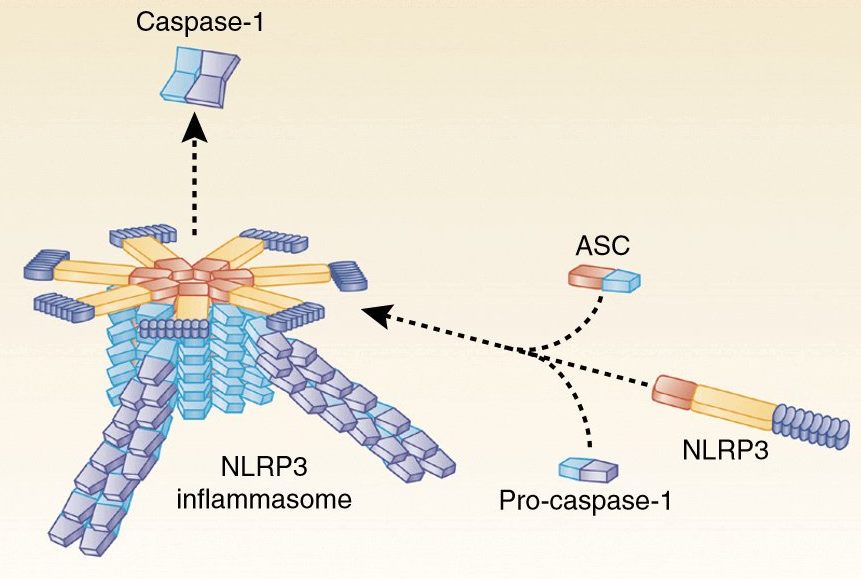
The lymphatic system is a network of thin tubes and nodes that runs throughout our bodies. The tubes are called lymphatic vessels, and they contain a liquid called (surprise!) lymph.
 The human lymphatic system. Source: Wikipedia
The human lymphatic system. Source: Wikipedia
The lymphatic system is similar to our blood circulation, with lymphatic vessels branching throughout our bodies like the arteries and veins.
The lymphatic system has two main functions:
- Fighting infections – lymph contains a type of white blood cells called lymphocytes, which help to fight infections and kill rogue cells
- Remove waste -the lymphatic system serves as a drainage system for the body, collecting waste products and excess fluid from all of the tissues and organs
In addition to the lymphatic vessels, there are also the lymph nodes. These are small bean-shaped glands, scattered throughout the body, which filter the lymph fluid as it passes through them. White blood cells within the lymph node attack any viruses or bacteria that they find in the lymph. One can imagine some pretty epic battles in lymph nodes.
 A lymph node. Source: CRUK
A lymph node. Source: CRUK
The big take away message is that the lymphatic system is critical to keeping our bodies healthy and free of waste, and any dysfunction could have serious consequences.
So the researchers wanted to know if there was any lymphatic dysfunction in this mouse model of Alzheimer’s? What did they find?
At 5-6 months of age, the 5xFAD mice displayed no evidence of lymphatic dysfunction. Everything looked like it was working ok.
 Source: PBS
Source: PBS
But by 13-14 months, the mice exhibited significant deterioration of the lymphatic vasculature in certain areas of the brain, and this was was associated with an increase in the aggregation of the Alzheimer’s-associated protein, beta-amyloid.
Next, the researchers asked if the changes in the lymphatic system would affect the ability of immunotherapy to clear the accumulated beta-amyloid protein. To test this, they injected the mice with two beta-amyloid-targeting antibodies, the mouse version of BAN2401 and Aducanumab (yes, that Aducanumab!).
 Source: Medpagetoday
Source: Medpagetoday
The scientists took young 5xFAD mice and artificially blocked the lymphatic system of half of them. They then treated all of the mice with the immunotherapies. While BAN2401 and Aducanumab treatment reduced the levels of beta-amyloid protein in all of the 5xFAD mice, the mice with blocked lymphatic systems presented larger amounts of accumulated beta-amyloid protein. In addition, there were also higher levels of neuroinflammation in these mice and they displayed worse performance in a test of memory (Morris water maze).
And the researchers reported that prolonged treatment with BAN2401 and Aducanumab did not improve the memory test performance.
The data suggested to the researchers that “lymphatic dysfunction accelerates cognitive decline and affects the removal of beta-amyloid from the brain by monoclonal antibodies“. They also found that microglia – the resident immune cells were affected by the lymphatic dysfunction.
They next explored whether VEGF-C treatment might be able to help improve the immunotherapy treatment.
What is VEGF-C?
Vascular endothelial growth factor C (or VEGF-C) is a growth factor – a protein that supports and nurtures cell growth. VEGF-C has been reported to enhance lymphatic drainage from the brain, AND this was associated with improved performance on learning and memory tests (Click here to read more about this).
When the researchers treated the 5xFAD mice mice with the BAN2401 immunotherapy AND a virus that causes cells to produce VEGF-C, they reported improvements in both the levels of beta amyloid in the brain as well as the function of the microglia.
The researchers concluded their study by suggesting that “the advanced stage of disease (or simply the advanced age) at which antibody-based therapies are administered might explain part of their marginal beneficial effects (and potential deleterious side effects), which could be attributed to compromised meningeal lymphatic function“. They propose that further research is required to identify biomarkers of lymphatic function, which could help facilitate a more personalised treatment approach.
Interesting. But what does this report have to do with Parkinson’s?
Given that the research has recently been published that indicates lymphatic dysfunction in some cases of Parkinson’s (Click here to read a recent SoPD post on that report), and antibody-based immunotherapy is being clinically tested in PD, the findings of this second study could have possible implications of Parkinson’s – it is certainly worth exploring.
|
# # RECAP #2: The lymphatic system is a network of thin tubes and nodes that runs throughout our bodies. It is used to fight infection and clear waste. New research suggests that in a mouse model of Alzheimer’s lymphatic dysfunction occurs and this could complicate the clearance of aggregated protein by immunotherapy approaches. # # |
So there are clinical trials for immunotherapy in Parkinson’s?
Yes, there are.
Quite a few actually. And all of them are trying to target the aggregated form of alpha synuclein protein.
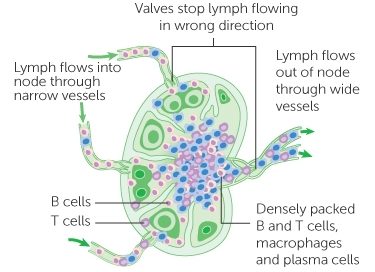
In 2020, the results of the PASADENA study were announced. This study was a Phase II clinical trial involving 316 individuals with recently diagnosed Parkinson’s being treated monthly with an alpha synuclein targeting immunotherapy (called Prasinezumab – formerly called RO7046015 & PRX002). The study was conducted by the pharmaceutical company Roche and biotech firm Prothena Biosciences (Click here to read a SoPD post about this).

In April 2020, the companies announced that the trial had not met its primary endpoint (a predetermined measure of efficacy), but that prasinezumab “showed signals of efficacy” , and importantly: “These signals were observed on multiple prespecified secondary and exploratory clinical endpoints“.
Specifically, when the researchers looked at just the motor scores of the participants (UPDRS Part III), there was evidence of a slower progression in the participants treated with prasinezumab than those treated with placebo (remember, these individuals were all blind to their treatment):
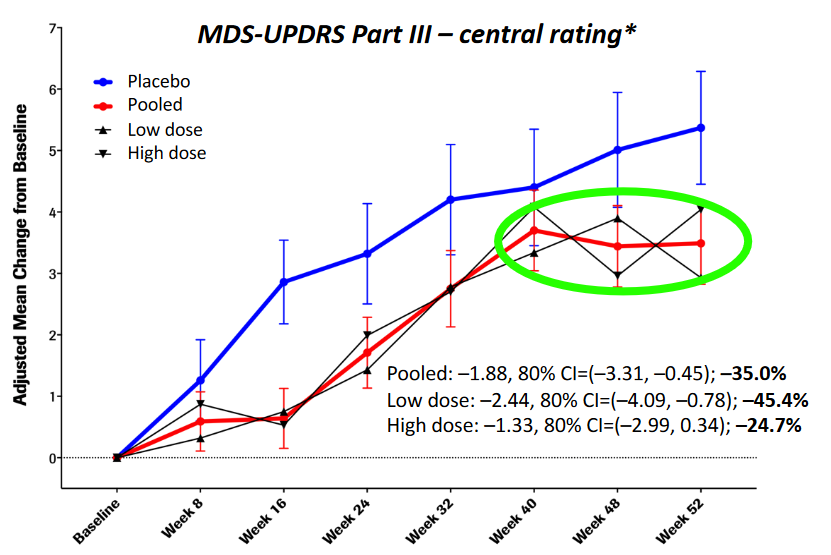 Source: Prothena
Source: Prothena
Roche and Prothena concluded that their “findings support the potential of prasinezumab to slow underlying disease pathophysiology and clinical decline in patients with PD. Further investigations are warranted” (Source). The two companies are continuing to follow up participant in the current Phase II study, and they have also initiated a Phase IIb clinical trial evaluating the efficacy and safety of Prasinezumab over 76 weeks of monthly treatment in in 575 individuals with recently diagnosed Parkinson’s.
This new trial is called the PADOVA study (Click here to read more about this trial).
That sounds positive. Are other biotech companies also testing immunotherapies in Parkinson’s?
It is encouraging.
But it is very important to remember that the positive findings of the Pasadena study are based on post hoc analysis (that is, after-the-fact re-analysis of the trial data) and as such they should not be considered as evidence of efficacy.
It would be even more encouraging if additional immunotherapy trials show signals that also warrant further investigation.
Speaking of which…
The second main immunotherapy programme for Parkinson’s was the SPARK study, that was conducted by the Pharmaceutical company Biogen (note the use of past tense).
 This was a 2-year Phase II clinical trial testing Biogen’s alpha synuclein targeting immunotherapy treatment BIIB054 (also known as Cinpanemab) in a group of 311 people with Parkinson’s. In the first year of the study, participants in the study were randomly assigned to monthly infusions of 3 different doses of BIIB054 (250mg, 1250mg, or 3500mg) or a placebo treated group (Click here to read more about this study and click here to read a SoPD post about the Phase I Biogen study results).
This was a 2-year Phase II clinical trial testing Biogen’s alpha synuclein targeting immunotherapy treatment BIIB054 (also known as Cinpanemab) in a group of 311 people with Parkinson’s. In the first year of the study, participants in the study were randomly assigned to monthly infusions of 3 different doses of BIIB054 (250mg, 1250mg, or 3500mg) or a placebo treated group (Click here to read more about this study and click here to read a SoPD post about the Phase I Biogen study results).
 In February of this year, Biogen announced to its investors that they had halted development of cinpanemab after the SPARK study missed both its primary and secondary endpoints (Click here to read more about this).
In February of this year, Biogen announced to its investors that they had halted development of cinpanemab after the SPARK study missed both its primary and secondary endpoints (Click here to read more about this).
That doesn’t sound encouraging. Are any other companies testing other immunotherapies for PD?
The results of the SPARK study have not yet been published, so it is difficult to say anything about the results. And perhaps Biogen will find some interesting trends in their post-hoc analysis of the data which will re-invigorate their interest (cough – aducanumab – cough).
And fear not, in addition to these two advanced immunotherapy programmes, there are a number of other biotech companies developing immunotherapy approaches for Parkinson’s, including:
- Astrazeneca‘s immunotherapy treatment called MEDI1341 (being developed with Takeda Pharmaceutical) is currently in Phase I safety testing in healthy volunteers. This study is scheduled to finish in April 2021 – thus we should get some news/update regarding this treatment in 2021 (Click here to read more about that study). In addition, in June 2020, Astra registered a second Phase I study assessing multiple ascending doses of MEDI1341 in people with Parkinson’s. That new study is scheduled to complete in July 2022 (Click here to read more about this trial).
- Lundbeck‘s immunotherapy treatment called Lu AF82422 (which is being developed in collaboration with Genmab) was in Phase I safety testing in both healthy volunteers and people with Parkinson’s during 2020 and it is scheduled to finish in August 2021 (Click here to read more about this).
- In March 2020, the pharmaceutical company AbbVie started a multicenter, placebo-controlled Phase I study of their immunotherapy treatment called BAN0805/ABBV-0805 (Click here to read more about this – this immunotherapy approach is being developed in collaboration with BioArctic Neuroscience). In June 2020, however, the Phase I study was withdrawn and BioArctic announced that a detailed plan to accelerate ABBV-0805 into a Phase II Proof of Concept study in Parkinson’s is now being prepared by AbbVie (Source) – we are yet to learn about that plan,, but the antibody is still listed in their pipeline.
- Biotech firm Scineuro recently announced a licensing deal with pharma company Eli Lilly for their alpha synuclein-targeting immunotherapies. Scineuro will develop these within Greater China, while Lilly will retains all rights outside of China.
So there is a lot clinical trial activity regarding immunotherapies for Parkinson’s.
But what about the two reports we have reviewed in today’s post? What are the implications of the results for these trials?
Difficult to say. Both studies need to be independently validated before we start making conclusions.
It could be, however, that some of these immunotherapies may become part of a combination therapy that includes some kind of anti-inflammatory or lymphatic system modulator.
Are there any clinical trial efforts focused on improving lymphatic flow?
Not that I am aware of (and I am happy to be corrected on this).
But there is a biotech company called PureTech Health that is exploring this area.
 PureTech have been developing a drug called deupirfenidone (previously known as LYT-100), which has anti-inflammatory, antioxidant and antifibrotic properties, but it is being targeted towards disorders of lymphatic flow (specifically lymphedema). It is also being targeted at Long COVID (Source).
PureTech have been developing a drug called deupirfenidone (previously known as LYT-100), which has anti-inflammatory, antioxidant and antifibrotic properties, but it is being targeted towards disorders of lymphatic flow (specifically lymphedema). It is also being targeted at Long COVID (Source).
The company is also exploring ways of delivering therapeutics via the lymphatic system. They are developing an agent, LYT-300, which is an oral form of FDA-approved allopregnanolone (a natural neurosteroid) for a range of neurological conditions. It is looking to harness “the role of the lymphatic system in the absorption of dietary lipids to orally administer and traffic therapeutics via the lymphatic system” (Source).
Perhaps this molecule could be part of a combination therapy approach involving immunotherapies targeted at Alzheimer’s and Parkinson’s?
So what does it all mean?
For anyone that has been following the immunotherapy field over time, they will know that it has been a wild rollercoaster. Some spectacular successes in oncology, and some painful disappointments in other areas (like Alzheimer’s). New insights could be helping to make the development of immunotherapies for neurodegenerative conditions a smoother ride.
Recently, one research group reported that some immunotherapies may be having the unintended consequence of enhancing inflammation, which could be negatively impacting the outcome of the treatment. Another research group has highlighted issues with the possible clearance of the targeted proteins.
As I said above, we need to see independent validation of both studies before we can really start drawing any conclusions. In the meantime, it could be interesting to look at inflammatory markers in blood or cerebrospinal fluid from some of the current immunotherapy studies to see if they are noting any increase.
It’s just a thought.
 All of the material on this website is licensed under a
All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
The banner for today’s post was sourced from

LikeLike
Simon,
Here is the link to an interesting publication:
https://www.medscape.com/viewarticle/953760?src=mkm_covid_update_210625_MSCPEDIT&uac=312734EZ&impID=3467714&faf=1
This publication describes changes in the brain tissue of people who died after contracting COVID19 virus. Here is a small excerpt from the article:
“The most comprehensive molecular study to date of brain tissue from people who died of COVID-19 provides clear evidence that SARS-CoV-2 causes profound molecular changes in the brain, despite no molecular trace of the virus in brain tissue.
“The signature the virus leaves in the brain speaks of strong inflammation and disrupted brain circuits and resembles signatures the field has observed in Alzheimer’s or other neurodegenerative diseases,” senior author Tony Wyss-Coray, PhD, professor of neurology and neurological sciences, Stanford University School of Medicine, Stanford, California, told Medscape Medical News.”
Is this another possible mechanism for PD start?
LikeLike
I’m not a researcher, but could anything be more obvious than that a therapy that incites the immune system to attack *anything* will lead to an in increase in inflammation?
And given that autoimmune inflammation is a central feature of Parkinson’s progression, how could anything that increases, rather than calms, microglia be of general benefit to a patient, no matter how much alpha-synuclein it removes?
The autoimmune process in PD is not just driven by aggregated alpha-synuclein. It is also driven by the presence of neuromelanin, released by damaged and dying neurons, in the intercellular space. Neuromelanin triggers receptors on microglia, just as aggregated alpha-synuclein does, causing them to release cytokines that damage previously-undamaged bystander neurons, which release more neuromelanin (and alpha-synuclein), completing the feedback loop.
So of course monoclonal antibodies that incite microglia to attack will feed this self-destructive process.
OTOH, we have substances like curcumin, baicalin, EGCg and quercetin that are capable of preventing, and even reversing somewhat, the aggregation of alpha-synuclein, while at the same time having a *calming* effect on microglia. Why not use these over-the-counter, inexpensive, non-proprietary and at the very least non-harmful means to counteract AS aggregation? In that last sentence, I may have just answered my own question.
LikeLike
I think I may have misunderstood something fundamental about this approach. It may be that the monoclonal antibodies are attempting to incite an adaptive immune response, and they have inadvertently triggered an innate response from the microglia due to the combination of the antibody with the aggregated AS protein. And perhaps it was not unreasonable to think that the adaptive response would be highly targeted to the AS protein, and thus would not stimulate a microglial response or otherwise harm nearby neurons. In which case, one might think that it was just bad luck that the combination of the antibody with the aggregated AS excited the microglia to further acts of destruction, and even allow for the possibility that a different antibody might not have that effect.
My remarks remain valid regarding the inflammatory feedback loop being sustainable even in the absence of aggregated AS (e.g., through the release of neuromelanin). And also, my remarks regarding the advantages of natural disaggregators of AS, and their simultaneous inhibition of microglial receptors.
But I will have to allow that if the problem of microglial reaction to the anti-body/protein complex could be resolved, this approach might, in theory, be somewhat helpful by reducing one important factor promoting inflammation and apoptosis.
LikeLike
Having made the above correction, I’d nonetheless note that it seems possible that the adaptive immune cells could also stimulate an innate response, and thus exacerbate inflammation, even in the absence of direct microglial pattern recognition receptor stimulation by an unfortunate antibody/AS complex. “T cells have the capacity to regulate and enhance the generation of early innate inflammatory responses within tissues upon cognate recognition of antigen. The antigen-specific regulation of inflammatory responses provides an additional means by which the immune response can generate alarm signals during infections with pathogens that possess means to evade detection by the innate immune-sensing mechanisms discussed earlier.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6016164/
LikeLike
From the same source: “An often-overlooked effector function of memory CD4 and CD8 T cells is the promotion of an inflammatory milieu at the initial site of infection that mirrors the primary encounter.”
LikeLike
Hi Lou,
One of the fundamental assumptions being made in a lot of this research is that alpha synuclein aggregation is the bad guy and that by being passed from cell to cell it is progressing the condition. I suspect that the jury is still out on this. The preclinical models used produce ridiculously high levels of protein aggregation, which might not be disease relevant. In addition, there is a fierce debate regarding gain-of-(toxic)-function vs loss-of-(physiological)-function surrounding raging in academic circles surrounding a-syn. I suspect that this line of thinking is too simplistic – a-syn is not the only protein that is aggregating in many cases of PD. This again points towards the need for a multi-modal approach towards therapy.
Kind regards,
Simon
LikeLiked by 1 person
In support of the ability of the supplements I have listed in my original comment to prevent aggregation of alpha-synuclein, and even to disaggregate such aggregates after they have already formed, we have the following review article:
https://www.frontiersin.org/article/10.3389/fphar.2018.01555/full
“Ono and Yamada (2006) found that curcumin possesses anti-fibrillogenic activity by inhibiting α-syn fibril formation and destabilizing preformed fibrils (Ono and Yamada, 2006). It was found to inhibit oligomerization of mutant α-syn into higher molecular weight aggregates (Pandey et al., 2008) and induce the dissociation of α-syn fibrils (Shoval et al., 2008). Curcumin treatment on mesencephalic cells did not affect α-syn fibril formation but enhanced LRRK2 mRNA and protein expression in rats (Ortiz-Ortiz et al., 2010). In neuroblastoma cells, curcumin attenuates cytotoxicity from aggregated α-syn, ROS generation, and diminished caspase-3 activation (Wang et al., 2010). In PC12 cells, curcumin ameliorates A53T mutant α-syn-induced PD (Liu et al., 2011). Further, curcumin reduces mutant α-syn accumulation by restoring macroautophagy, a process in the degradation pathway that clears proteins in cells by activating the mTOR/p70S6K signaling pathway (Jiang et al., 2013). Mechanistically, curcumin preferentially binds oligomeric intermediates rather than monomeric α-syn (Singh et al., 2013). Also, it binds strongly to the hydrophobic non-amyloid-β component of α-syn (Ahmad and Lapidus, 2012). The ordered structure is vital for effective binding and affects the extent of binding and potential in inhibiting oligomers or fibrils (Singh et al., 2013). The conformational and reconfiguration changes appear to govern the binding of curcumin to α-syn (Ahmad and Lapidus, 2012; Tavassoly et al., 2014).
“EGCG inhibited α-syn aggregation and fibrillation in a concentration-dependent manner (Šneideris et al., 2015; Xu et al., 2017), and by disaggregating mature and large α-syn fibrils into smaller, non-toxic, amorphous aggregates (Ehrnhoefer et al., 2008). EGCG binds directly to the natively unfolded polypeptides and inhibits their conversion into toxic intermediates (Ehrnhoefer et al., 2008). It induces a conformational change without their disassembly into monomers or small diffusible oligomers (Bieschke et al., 2010). It appears to bind directly with β-sheet-rich aggregates and reduces its concentration required to induce conformational changes (Liu et al., 2018). Furthermore, it showed neuroprotection against free radicals and α-syn toxicity by chelating Fe (III) in PC12 cells transfected with α-syn and exposed to β-sheet-enriched α-syn fibrils (Zhao et al., 2017). EGCG appears to disaggregate α-syn fibrils by preventing the amyloid formation of α-syn tandem repeat and destabilizing α-syn fibrils into soluble amorphous aggregates (Bae et al., 2010).
“Recently, in primary cortical neuron cultures challenged with oxidative injury, quercetin, EGCG, and cyanidin-3-glucoside inhibited fibrillation of α-syn and apoptosis (Pogacnik et al., 2016). Further, it decreased amyloid fibril formation on the surface of liposomal membranes and generates compact oligomers following off-pathway, as well as facilitating the conversion of active oligomers into amyloid fibrils (Yang et al., 2017). A combination of EGCG with specific α-syn proteolytic peptide sequences was developed for targeted drug delivery and found to prevent the α-syn fibrillation (Yoshida et al., 2013). In combination, this evidence suggests EGCG could be a promising treatment in neurodegenerative diseases and a good candidate for pharmaceutical development and dietary inclusion.
“In many studies, baicalein was shown to prevent α-syn oligomerization and fibrillation (Bomhoff et al., 2006; Meng et al., 2009; Caruana et al., 2011; Gasiorowski et al., 2011; Sashourpour et al., 2017). Baicalein interacts with α-syn through a tyrosine residue. Following oxidation, it generates quinone metabolites that bind covalently with a lysine side chain in α-syn. It prevents fibril formation and degrades preformed fibrils at low micromolar concentrations (Zhu et al., 2004). In another study, its non-covalent binding with α-syn and covalent modification by the oxidized form restricts the conformational changes in the unfolded protein that results in α-syn monomer and oligomer stabilization (Meng et al., 2009)…In a recent study, baicalein induces autophagy, increases cell viability and reduces α-syn in the media of dopaminergic cell lines (SN4741) overexpressing A53T-syn (Li et al., 2017). Baicalein diminished the transmission of α-syn and prompted the polymerization of α-syn to a big complex rather than promoting clearance (Li et al., 2017).”
LikeLike
Hi Lou,
Thanks for the (always) interesting comment. One of the issues with curcumin, baicalin, EGCg and quercetin is that they all have poor bioavailability and elimination half lives. Maybe we don’t need too much, I am not sure, but this is why nanoparticle reformulations (like Theracurmin) have been generated.
In addition, the results of the PROMESA study (a Phase II clinical trial of EGCG in multiple systems atrophy – a more likely synucleinopathy than PD) indicate that this therapy had no impact on progression (https://pubmed.ncbi.nlm.nih.gov/31278067/).
I think the real lesson from the two papers reviewed in this post is that the ‘monotherapy’ approach may be too simplistic.
Kind regards,
Simon
LikeLiked by 1 person
Yes, we are using a nanoparticle formulation developed at UCLA called Longvida curcumin, which has greatly enhanced bioavailability. The nanoparticles are encapsulated in lipid shells that prevent the liver from tagging the curcumin particles for elimination via the glucuronidation process. This results in their hanging around for a much greater length of time.
I agree with you that intervention with multiple synergistic substances is more likely to yield good results than will the use of any one substance. Unfortunately, most studies attempt to isolate the effects of a single substance, since otherwise the space being explored by the study can become unmanageable.
My own informal model is that there is an autoimmune feedback loop that can be damped down by making inhibitory interventions at multiple points in its cycle of causation. So some of the substances like curcumin are anti-inflammatories that inhibit receptors (like TLR2 and 4) on microglia, and others are antioxidants (like EGCg and NAC) that interfere with the cascade that triggers neurona death in response to cytokines that microglia emit, and some substances (like ubiquinol) support mitochondrial function so that neurons can better defend themselves against attack. At a certain point, the hope is that the feedback loop is damped down enough that it peters out. Because feedback loops have a threshold and if you can get even a tiny bit below it, they may suddenly stop.
That gives a specific version of the synergy we are talking about, and shows why monotherapies may fail and yet could be part of a larger strategy involving multiple substances that could succeed.
LikeLike