|
There is a lot of clinical and biological similarities between the neurodegenerative conditions of Parkinson’s and multiple systems atrophy (or MSA). Recently, however, researchers have published a report suggesting that these two conditions may be differentiated from each other using a technique analysing protein in the cerebrospinal fluid – the liquid surrounding the brain, that can be accessed via a lumbar puncture. Specifically, the method differentiates between different forms of a protein called alpha synuclein, which is associated with both conditions. In today’s post, we will look at what multiple systems atrophy (MSA) is, discuss how this differentiating technique works, and explore what it could mean for people with either of these conditions.
|
 Source: Assessment
Source: Assessment
Getting a diagnosis of Parkinson’s can be a tricky thing.
For many members of the affected community, it is a long and protracted process.
Firstly, there will be multiple visits with doctors and neurologists (and perhaps some brain imaging) until one is finally given a diagnosis of PD. There are a number of conditions that look very similar to Parkinson’s, which must be ruled out before a definitive diagnosis can be proposed.
But even after being diagnosed, there are a group of conditions that look almost identical to Parkinson’s. And many people will be given a diagnosis of Parkinson’s before they are then given a corrected diagnosis of one of these other conditions.
Can you give me an example of one of these other conditions?
Sure. A good example is multiple systems atrophy.
What is Multiple System Atrophy?
Multiple System Atrophy (MSA), also known as Shy-Drager syndrome, is a rare neurodegenerative condition. It is considered an Atypical Parkinsonism.
What is an ‘Atypical Parkinsonism’?
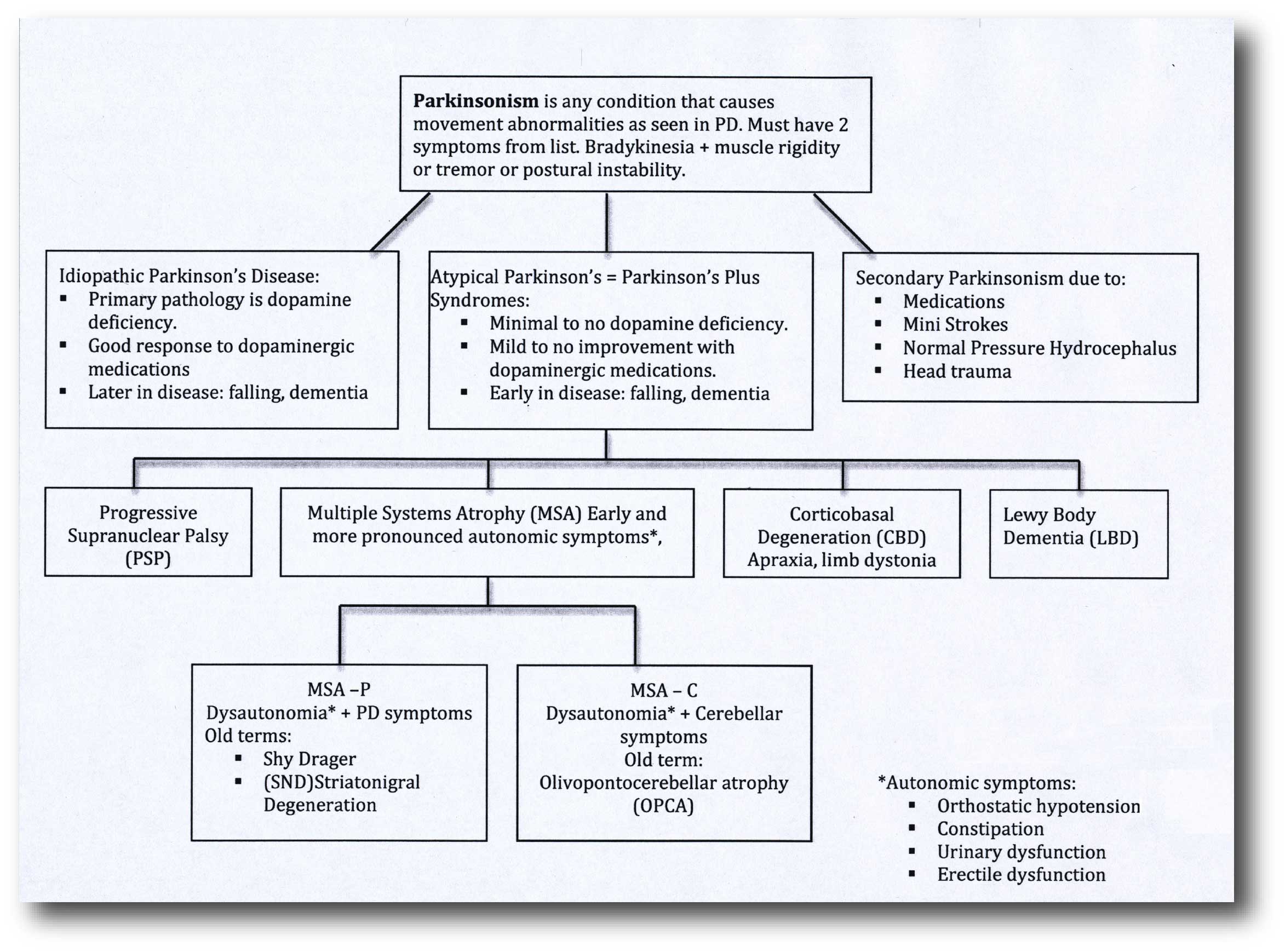
‘Parkinsonisms’ refer to a group of neurological conditions that cause movement features similar to those observed in Parkinson’s disease, such as tremors, slow movement and stiffness. The name ‘Parkinsonisms’ is often used as an umbrella term that covers Parkinson’s disease and all of the other ‘Parkinsonisms’.
Parkinsonisms are generally divided into three groups:
- Classical idiopathic Parkinson’s (the common spontaneous form of the condition)
- Atypical Parkinson’s or Parkinson-plus syndromes (such as multiple system atrophy and Progressive supranuclear palsy (PSP))
- Secondary Parkinson’s (which can be brought on by mini strokes (aka Vascular Parkinson’s), drugs, head trauma, etc)
 Source: Parkinsonspt
Source: Parkinsonspt
Some forms of Parkinsonisms that at associated with genetic risk factors, such as juvenile onset Parkinson’s, are considered atypical. But as our understanding of the genetics risk factors increases, we may find that an increasing number of idiopathic Parkinson’s cases have an underlying genetic component (especially where there is a long family history of the condition) which could alter the structure of our list of Parkinsonisms.
So what is the difference between Multiple system atrophy and idiopathic Parkinson’s?
When a person first presents at the clinic with rigidity, slowness of movement and a resting tremor, it can be very difficult to differentiate between classical idiopathic Parkinson’s and other kinds of Parkinsonisms. But there are some telltale signs that can help differentiate the Multiple System Atrophy from idiopathic Parkinson’s:
- The disability progresses more rapidly in MSA
- People with MSA are poor responders to levodopa treatment over time (such as Sinemet)
- Urinary retention and orthostatic hypotension (blood pressure falls when suddenly standing up) are common
- Rigidity and bradykinesia are out of proportion to tremor
- Speech is severely affected
- Gasping and high pitched wheezing sounds when breathing are present
- In the vast majority of cases, there is an absence of dementia, though other cognitive functions can be affected
- Reduced blinking and dry eye, in addition to jerky or slower eye movements
And the Second Consensus Statement on the diagnosis of multiple system atrophy provides for two basic types of multiple system atrophy, based on the symptoms of the condition at the time of evaluation.
These two types are:
- MSA with predominant parkinsonism (MSA-P), which can resemble idiopathic Parkinson’s because of slow movement and stiff muscles. The terms striatonigral degeneration or parkinsonian variant are sometimes used for this category of MSA.
- MSA with cerebellar features (MSA-C). It is sometimes termed ‘sporadic olivopontocerebellar atrophy’ (or OPCA). MSA-C primarily affects balance, coordination, and speech.
Some researchers believe that there is also a third type of MSA which is a combination of the two.
How is Multiple system atrophy diagnosed?
Currently there is no test that can definitively make or confirm the diagnosis of MSA in a living person.
Having said that, regular use of brain imaging techniques such as MRI and CT scanning can show a decrease in the size of specific brain structures affected in MSA (such as the cerebellum and pons). For example, in the image below you can see serial MRI images of the brain of a person with MSA-C over a 2-year period. Note the subtle reduction in the size of the characteristic ‘hot cross bun’ shape of the pons between image C (2004) and two years in image D (2006):
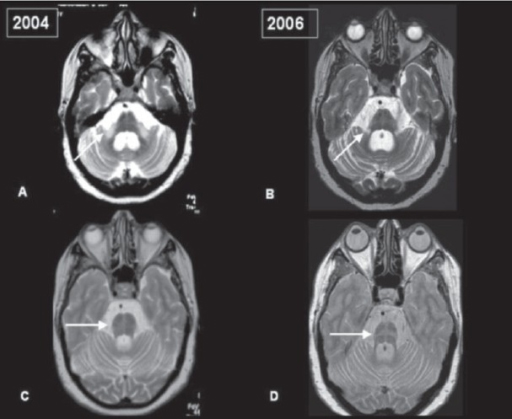 Serial MRI of brain of a person with MSA-C over a 2-year period. Source: Openi
Serial MRI of brain of a person with MSA-C over a 2-year period. Source: Openi
And in MRI brain scan images from individuals affected by MSA-P, there is a gradual darkening of the region of the brain called the putamen (indicated on the left side of the brain by a white arrow in the image below). You can see that over time as the condition progresses (from grade 0 to grade 3), the putamen becomes darker and darker.
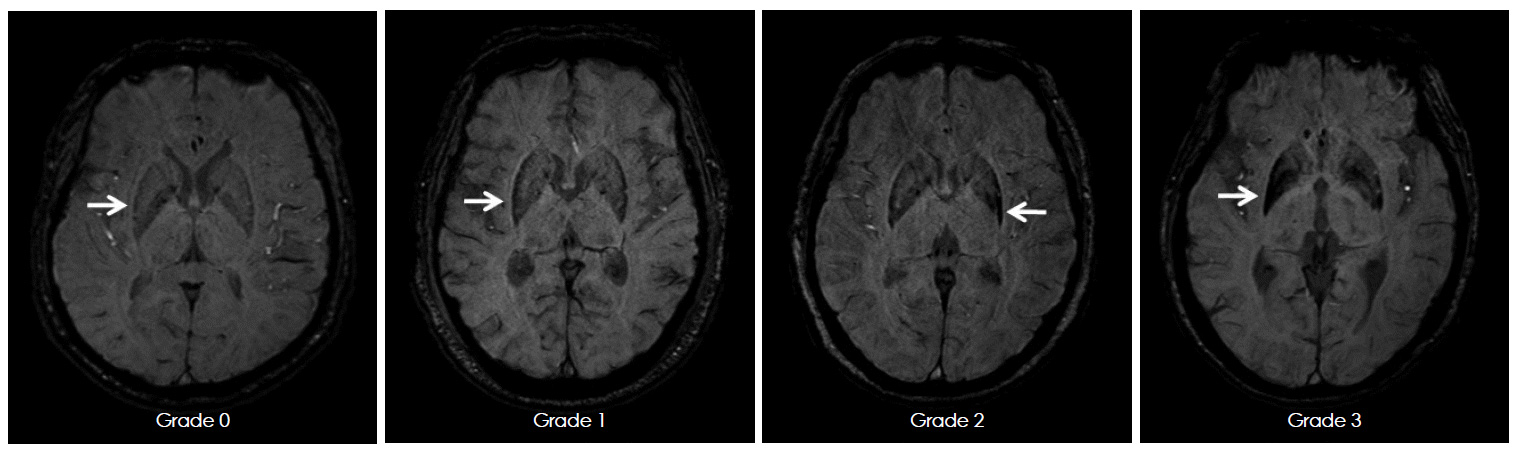 MRI of brain of a person with MSA-P over time. Source: e-jmd
MRI of brain of a person with MSA-P over time. Source: e-jmd
|
RECAP #1: Multiple systems atrophy (or MSA) is a neurodegenerative condition that is similar to Parkinson’s, but is considered an atypical form. It is associated with a faster rate of progression, poorer response to levodopa treatment, and more affected speech. |
What causes Multiple system atrophy?
I think most researchers would agree that the correct answer to this question is:
 Source: Wellbeing365
Source: Wellbeing365
Having said that, there is now a growing pile of evidence that a prion form of the Parkinson’s associated protein alpha synuclein may have a major influence in MSA.
What does ‘prion’ mean?
A prion is an infectious agent that is entirely protein based.
What does that mean?
Most infectious agents (like viruses or bacteria) are complex, and involve the transfer of DNA and RNA in order for replication or damage to be done.
In the case of a prion, there is no DNA/RNA transfer. Prions are simply proteins that have gone rogue.
Gone rogue?
Proteins ‘go rogue’ when they don’t fold properly. In order to do their particular job correctly, each protein must fold itself in particular ways to achieve a specific shape. Proteins are tiny machines and each type of protein has a different shape (many of them have several different shapes that they can adopt). The take-home message, however, is that if they do not fold correctly, they can not do their job.
For a good explanation of protein folding, watch this video:
Prions are proteins that present alternative configurations to their normal shape, but the critical distinguishing feature of prions is that the protein becomes self-propagating. That is to say, the alternative folding is transmissible to other prion-prone proteins. One protein that has gone rogue (and become a prion) can encourage another protein to go rogue. And this ultimately leads to a cascade that very much resemble the spread of a viral infection.
How does this work?
The basic idea is that a Prion prone Protein (or PrP – see the image below) will sit inside a cell having various functions which it conducts in a normal set of folded configurations.
This normal state of the protein is called PrPC (C refers to ‘cellular’). Then one of two things could happen:
- the PrPC will spontaneous take on the infectious form of the protein, which is called PrPSc (Sc refers to ‘scrapie’, the prototypic prion disease occurring in sheep); or
- the PrPC will bump into a PrPSc which will then induce the PrPC to fold into the PrPSc configuration, thus causing the disease to spread.
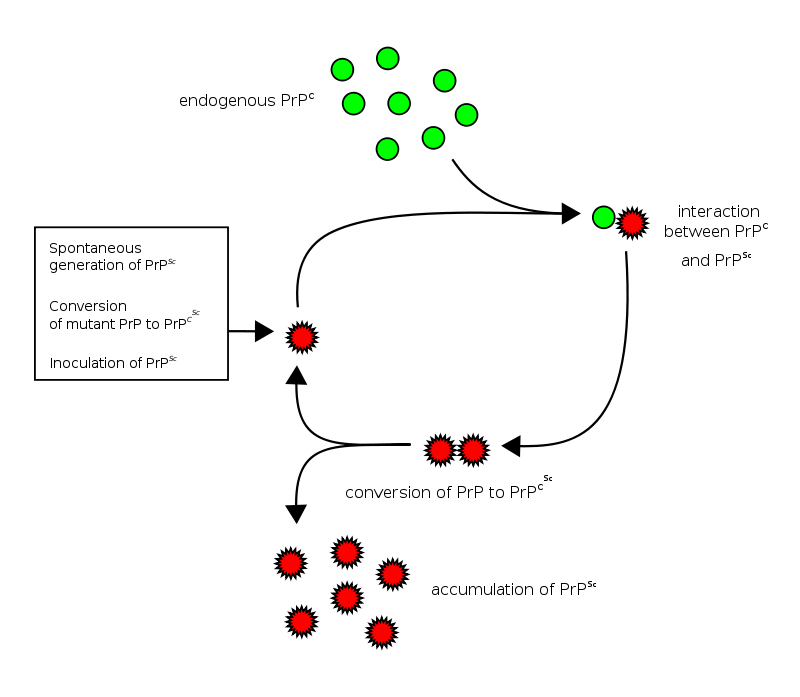 Prion life-cycle. Source: Wikipedia
Prion life-cycle. Source: Wikipedia
The most well researched example of a prion condition is Creutzfeldt-Jakob disease. This video will explain for you what CJD is:
So researchers think that alpha synuclein could be a prion-prone protein?
There is certainly evidence to suggest that alpha synuclein can act like a prion.
Remind me again, what is alpha synuclein?
When a protein is produced (by stringing together amino acids in a specific order set out by the RNA instructions), it will then be folded into a functional shape that do a particular job (as we have discussed above).
Alpha synuclein is slightly different in this respect. It is normally referred as a ‘natively unfolded protein’, in that is does not have a defined structure. Alone, it will look like this:
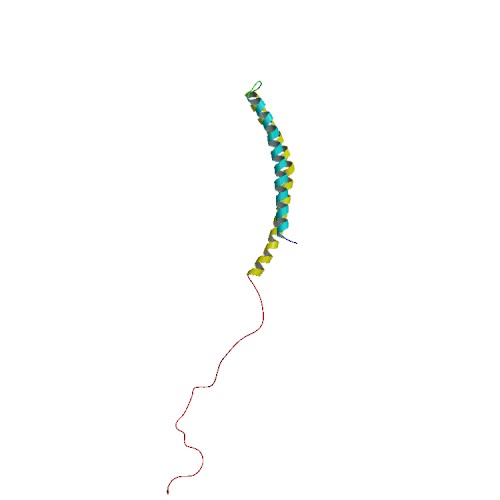 Alpha synuclein. Source: Wikipedia
Alpha synuclein. Source: Wikipedia
By itself, alpha synuclein is considered a monomer, or a single molecule that will bind to other molecules to form an oligomer (a collection of a certain number of monomers in a specific structure). In Parkinson’s disease, alpha-synuclein also aggregates to form what are called ‘fibrils’.

Microscopic images of Alpha Synuclein (AS) monomers, oligomers and fibrils. Source: Brain
Oligomer versions of alpha-synuclein are emerging as having a key role in Parkinson’s disease. They lead to the generation of fibrils and may cause damage by themselves.

Source: Nature
It is believed that the oligomer versions of alpha-synuclein is being passed between cells – and this is how the disease may be progressing – and once inside the cell, these oligomer versions of alpha synuclein are causing other ‘natively unfolded protein’ monomer versions of alpha synuclein to become oligomers, which either bind with other oligomers to form fibrils or move on to another cell.
Hopefully you can see why some researchers are suggesting that alpha synuclein has prion-like properties, causing a chain-reaction that gives rise to Parkinsonisms like MSA and idiopathic Parkinson’s.
 Is Alpha Synuclein a prion? Source: PNAS
Is Alpha Synuclein a prion? Source: PNAS
For a good review of the alpha synuclein prion theory – Click here.
|
RECAP #2: Alpha synuclein – which is associated with both Parkinson’s and multiple systems atrophy – may be a prion protein. That is a protein that goes rogue and acts as an infectious agent, changing other proteins like it into ‘prions’. |
Interesting. So what does this new report suggest?
This is the new study here:
 Title: Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy.
Title: Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy.
Authors: Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, Hu B, Schmeichel A, Singer W, Wu G, Tsai AL, Shirani H, Nilsson KPR, Low PA, Soto C.
Journal: Nature. 2020 Feb 5.
PMID: 32025029
In this study, the researchers used “protein misfolding cyclic amplification” to discriminate between samples of cerebrospinal fluid from people diagnosed with Parkinson’s and samples from people diagnosed with MSA.
What is “protein misfolding cyclic amplification”?
Protein misfolding cyclic amplification (or PMCA) is a method of amplifying ‘prion-like proteins’ and determining/measuring featuring of their prion-like behaviour.
The technique involves a small amount of a prion-like protein being incubated with a whole lot of normal protein. If the prion-like protein truly is prion-like, it will start converting the normal protein into prion proteins. To speed the process up, the growing chains of prion-like proteins is repeatedly blasted with ultrasound, which breaks the chain down into smaller chains. This rapidly increases the amount of prion-like protein available to cause further conversions. And by repeating the cycle multiple times, the bulk of the normal protein will rapidly be changed into the prion being investigated.
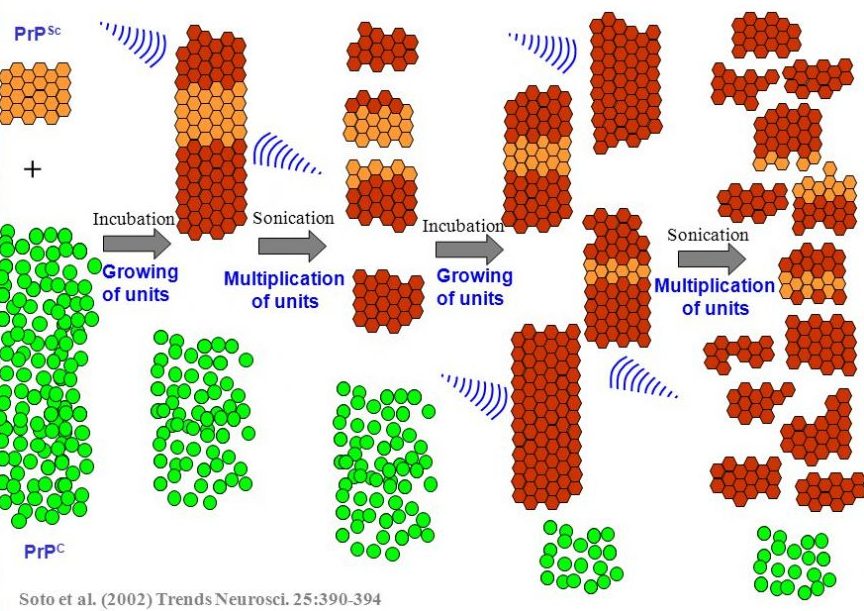 Source: Slideplayer
Source: Slideplayer
This approach was first proposed in 2001 and has become widely used to investigate proteins with prion-like properties:
 Title: Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding.
Title: Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding.
Authors: Saborio GP, Permanne B, Soto C.
Journal: Nature. 2001 Jun 14;411(6839):810-3.
PMID: 11459061
In the more recent study investigating samples of alpha synuclein from MSA and Parkinson’s patients, the investigators used the PMCA method on 94 samples of cerebrospinal fluid from people with Parkinson’s, 75 from people with MSA, and 56 from control individuals who had other neurological conditions.
The investigators found that the alpha synuclein ‘prions’ from Parkinson’s samples had a different ‘signature’ to those of the MSA samples. And they could accurately determine which disease each sample was coming from based on that signature.
For example, when the researchers looked at the cases of Parkinson’s, of the 88 samples of cerebrospinal fluid that showed alpha synuclein with prion-like behaviour, 85 were correctly identified as Parkinson’s (for the maths fans in the audience – that is an accuracy rate of 96.6%).
And for the 65 samples from MSA cases that exhibited alpha synuclein with prion-like behaviour, 61 had the signature typical of MSA (94%… mmm, not bad!). What’s more, if you combine all of the samples, the researchers correctly distinguished the Parkinson’s samples from the MSA samples in 146 of 153 cases (that’s 95%).

The researchers also demonstrated that the alpha synuclein aggregates that are present in the cerebrospinal fluid was representative of those collected from postmortem brain samples. While the number of samples analysed in this last analysis was much smaller, the finding is important as it suggests that the PMCA technique may be able to determine pathological forms of alpha synuclein that are associated with different synucleinopathies from a simple collection of spinal fluid (I say ‘simple’, but it can be a painful process – better than a brain biopsy though, I suppose).
And this means that we could be looking at the development of a biochemical test that could aid the diagnostic process and have serious implications for future clinical trials (both in terms of recruitment and novel therapies) as well as more personalised approaches to treatment.
|
RECAP #3: Researcher have used a method of amplifying alpha synuclein ‘prions’ to differentiate between Parkinson’s and multiple systems atrophy to an accruacy of 95%. This finding could have important implications regarding the development of a biochemical test to aid the diagnostic process, as well as clinical trials and more precise treatment in the future. |
So alpha synuclein aggregates differ between conditions?
That appears to be the case.
In their study, the researchers reported that the alpha synuclein aggregates that were associated with Parkinson’s and multiple system atrophy “corresponded to different conformational strains of alpha synuclein“.
And this idea is supported by another very recent manuscript that has been posted on the preprint website bioRxiv:
 Title: Structures of α-synuclein filaments from multiple system atrophy
Title: Structures of α-synuclein filaments from multiple system atrophy
Authors: Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, Murayama S, Yoshida M, Hasegawa M, Scheres SHW, Goedert M
Doi: https://doi.org/10.1101/2020.02.05.935619
In this manuscript, the researchers have presented data suggesting that the conformation (or shape) of alpha synuclein aggregates from the postmortem brains of people who passed away with multiple systems atrophy are different to those from the brain of a person who died with Lewy body dementia (Click here to read a previous SoPD post on LBD). Their results leave the investigator “suggesting that distinct conformers (or strains) characterise synucleinopathies“.
And one very important take away from this research is that the researchers also noted that the conformation of the alpha synuclein they were seeing in the multiple systems atrophy cases was different to that of the alpha synuclein being generated in laboratories and used in experiments.
What does that mean?
In labs, researchers will generate alpha synuclein aggregates ‘in test tubes’ to perform their experiments. These “preformed fibrils” of alpha synuclein are easy to generate compared to acquiring and extracting aggregates from human postmortem brain.
 Source: IOS
Source: IOS
BUT these new results may call into question if the “preformed fibrils” of alpha synuclein that are generated in the lab behave the same way as alpha synuclein extracted from the brain of a person who passed away with multiple systems atrophy (or Parkinson’s). And this situation is not unique to alpha synuclein. There have been similar reports for the neurodegeneration associated protein Tau (click here to read more about this).
More research is needed in this area.
Interesting. So what does this mean for Parkinson’s (and MSA)?
There are a large number of clinical trials focused on targeting alpha synuclein in both Parkinson’s and multiple systems atrophy (click here to read about some examples). Some of these studies involve boosting the immune system to grab alpha synuclein as it is floating around outside of cells (these are refered to as immunotherapy – click here to read a previous SoPD post on this topic)
There are also a number of studies focused on testing small molecules that can actually access neurons and disaggregate aggregated alpha synuclein.
One example of this is being explored by a company called MODAG.
 They are currently Phase 1 clinically testing their lead compound which is called Anle138b, with the aim of testing it in people with multiple systems atrophy (and hopefully also testing it in Parkinson’s – Click here to read a previous SoPD post on MODAG and Anle138b)
They are currently Phase 1 clinically testing their lead compound which is called Anle138b, with the aim of testing it in people with multiple systems atrophy (and hopefully also testing it in Parkinson’s – Click here to read a previous SoPD post on MODAG and Anle138b)
Another example is a drug called NPT088, which is being developed by Proclara Biosciences.
![]()
Formerly called NeuroPhage Pharmaceuticals, Proclara is clinically testing NPT 088 which can apparently break down any aggregated protein (beta amyloid, Tau, alpha synuclein, etc). It was being tested in individuals with probable Alzheimer’s (Click here to read more about this) – the company has stated on their website that the results would be applicable to Parkinson’s (Source). The trial finished in February 2019, and the results have recently been published (Click here to read more about this). In 2020, we are hopeful to hear news of further development of this drug.
And another biotech firm developing a small molecule to target alpha synuclein is NeuaroPore Therapies.

They are clinically testing a small molecule inhibitor of alpha synuclein called NPT520-34. Phase I testing in healthy individuals finished in September 2019, so we are also hopeful to hear the results of that study this year (Click here to read more about this trial).
NeuroPore Therapies has also out-licensed another small molecule alpha synuclein aggregation inhibitor called NPT200-11 to the pharma company UCB.

This drug has also been Phase I tested (Click here to read more about that trial). The study was completed (Click here to read the press release), and we look forward to learning more about the future development of this drug this year.
So what does it all mean?
Aggregated protein in the brain is a cardinal feature of many neurodegenerative conditions, including Parkinson’s, Alzheimer’s and multiple systems atrophy. Recent research suggests that we may be able to differentiate between conditions that exhibit aggregation of similar proteins by investigating the conformation of the aggregated protein. Specifically, researchers have demonstrated that the alpha synuclein-associated conditions Parkinson’s and multiple systems atrophy can be determined using cerebrospinal fluid samples (it will be interesting to investigate if blood sames can be used).
IF aggregating proteins like alpha synuclein are eventually found to have a causal role in the pathology associated with these conditions, the new findings discussed in today’s post (suggesting specific conformations of the protein differentiate the conditions) will have important implications for how we a.) diagnose, b.) track, and c.) treat the conditions.
That underlined ‘IF’ is a big IF though.
And then there is also the elephant in the room: what causes these specific conformations of alpha synuclein and other aggregating proteins.
But we’ll explore that is another post. Good night all.

All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
The banner for today’s post was sourced from Scientific American

Great post. Data, hope and realism in one thread. Many thanks.
Big IF indeed… can we sacrifice a few thousand mice? Give them two forms of alpha synuclein and see if the develop mouse MSA vs mouse Parkinson’s? If we got 95% correlation, it would strengthen the causality theory for both.
LikeLike
Simon
Thanks for this very good explanation of MSA. You do not mention the involvement of United Neurosciences UNS and their immunotherapy approach.
When will you be doing a similar post on other atypical parkinsonisms, partcularly PSP?
Hope you are not overdoing it with late nght postings.
K
LikeLike