This week the ‘Michael J. Fox Foundation for Parkinson’s Research’ and ‘The Silverstein Foundation for Parkinson’s with GBA’ announced that they are collaboratively awarding nearly US$3 million in research grants to fund studies investigating an enzyme called beta glucocerebrosidase (or GCase).
Why is this enzyme important to Parkinson’s?
In today’s post, we will discuss what GCase does, how it is associated with Parkinson’s, and review what some of these projects will be exploring.
He is a General Partner of Global Private Equity at OrbiMed – the world’s largest fully dedicated healthcare fund manager. During his time at OrbiMed, the company has invested in healthcare companies that have been involved with over 60 FDA approved products.
In February 2017 – at just 49 years of age – Jonathan was diagnosed with Parkinson’s.
Rather than simply accepting this diagnosis, however, Mr Silverstein decided to apply the skills that he has built over a long and successful career in funding biotech technology, and in March 2017, he and his wife, Natalie, set up the Silverstein Foundation for Parkinson’s with GBA.
The foundation has just one mission: “to actively pursue and invest in cutting edge research with the goal of discovering new therapies for the treatment of Parkinson’s Disease in GBA mutation carriers”
And it seeks to address this by achieving three goals:
to find a way to halt the progression of Parkinson’s with GBA.
to identify regenerative approaches to replace the damaged/lost cells
At 9am on the 30th January, 2019, the Australian Government Federal Health Minister Greg Hunt announced the initiation of the ‘Australian Parkinson’s Mission‘ – a very massive $30 million clinical trial programme that will be focused on potentially disease modifying treatments for Parkinson’s.
This huge endeavour will being with a large multi-arm study – involving 300 hundred participants and investigating 4 drugs (compared to a single placebo). It will be a first of its kind project in the world targeting Parkinson’s.
This is a very exciting development for the Parkinson’s community!
In today’s post, we will discuss what we currently know about the Australian Parkinson’s Mission project, what we hope to see resulting from the initiative, and why this is a tremendous step forward for the international Parkinson’s community as a whole.
Being a patriotic kiwi there is always enormous potential to make fun when writing a post about any Parkinson’s-related news coming out of Australia. New Zealand and Australia have always had a big brother/little brother kind of relationship (and just so we are clear: NZ is the big brother!).
But today is different.
It is very strange to say, but… today… I am actually very proud of you Australia.
At 9am this morning at the Garvan Institute of Medical Research in Sydney, Greg Hunt – the Federal Health Minister of the Australian Government – announced the commencement of a major clinical trial initiative (named ‘The Australian Parkinson’s Mission‘), which is going to be a very large, world-leading clinical programme focused on potentially disease modifying drugs for Parkinson’s (Click here to read the press release).
Struth mate!!! This sounds fantastic. What do we know about the study?
Numerous readers have asked about a curious new clinical trial being conducted by a biotech firm called ‘Alkahest’. The company has recently initiated a large (90 participants) Phase II study of their Parkinson’s-focused treatment called GRF6021.
This is an experimental, intravenously-administered treatment, which is derived from a components of blood.
In today’s post, we will discuss some of the research behind GRF6021, what this new clinical trial involves, and have a look at some other interesting Parkinson’s-related activities that Alkahest has ongoing.
The Society of Neuroscience meeting is the largest annual research conference on brain relelated research, bringing approximately 40,000 neuroscientists together in October. At the Society of Neuroscience meeting in San Diego this year, however, there was considerable interest focused on several presentations dealing with blood.
The first presentation was from a group of researchers at the University of California, San Francisco.
The research team – led by group leader Dr Saul Villeda – were presenting new data suggesting that circulating immune cells were most likely responsible for the age-related reduction in neurogenesis (formation of new neurons) that occurs in certain areas of the brain (Click here to read the abstract for this presentation). They reported that the aged hematopoietic (blood) system led to impaired neurogenesis. Their take-home-message: the older the blood system, the less new cells being produced by the brain.
Sounds interesting right?
Well, at the same time in another part of the conference a second group of researchers were presenting equally impressive data: They have zeroed in of a small fraction of normal, young blood that they believe has interesting properties, particularly in reversing the cognitive deficits associated with aging mice (Click here to read the abstract of this presentation).
Their research has even narrowed down to a specific protein, called C-C chemokine receptor type 3 (or CCR3), which when inhibited was found to improve cognitive function and decreased neuroinflammation in aged mice (Click here to read the abstract of the presentation).
But specifically for our interests here at the SoPD, these same researchers displayed data which demonstrated that treatment with a novel fraction of human plasma resulted in significant improvements in motor function, cell survival and neuroinflammation three weeks after treatment in multiple mouse models of Parkinson’s (Click here to read the abstract of the poster).
(PLEASE NOTE: The author of this blog was not present at the SFN meeting and is working solely with the abstracts provided)
This second group of scientists were from a company called Alkahest, and they have recently started a clinical trial for people with Parkinson’s based on these results. That trial has garnered quite a bit of interest in the Parkinson’s community.What do Alkahest do?
Today’s post involves massive multidimensional datasets, machine learning, and being able to predict the future.
Sound interesting?
Researchers are the National Institute on Aging and the University of Illinois at Urbana–Champaign have analysed longitudinal clinical data from the Parkinson’s Progression Marker Initiative (PPMI) and they have found three distinct disease subtypes with highly predictable progression rates.
NOTE: Reading about disease progression may be distressing for some readers, but please understand that this type of research is critical to helping us better understand Parkinson’s.
In today’s post, we will look at what the researchers found and discuss what this result could mean for the Parkinson’s community.
Today I am going to break one of the unwritten rules of science communication (again) .
Until a research report has been through the peer-review process you probably should not be discussing the results in the public domain.
But in this particular case, the research is really interesting. And it has been made available on the OPEN ACCESS preprint depository website called BioRxiv.
I should add that this is not the first time we have discussed manuscripts on BioRxiv (Click here and here to read other post on Biorxiv manuscripts). We are regular rule breakers here at the SoPD.
At the end of each year, it is a useful practise to review the triumphs (and failures) of the past 12 months. It is an exercise of putting everything into perspective.
2017 has been an incredible year for Parkinson’s research.
And while I appreciate that statements like that will not bring much comfort to those living with the condition, it is still important to consider and appreciate what has been achieved over the last 12 months.
In this post, we will try to provide a summary of the Parkinson’s-related research that has taken place in 2017 (Be warned: this is a VERY long post!)
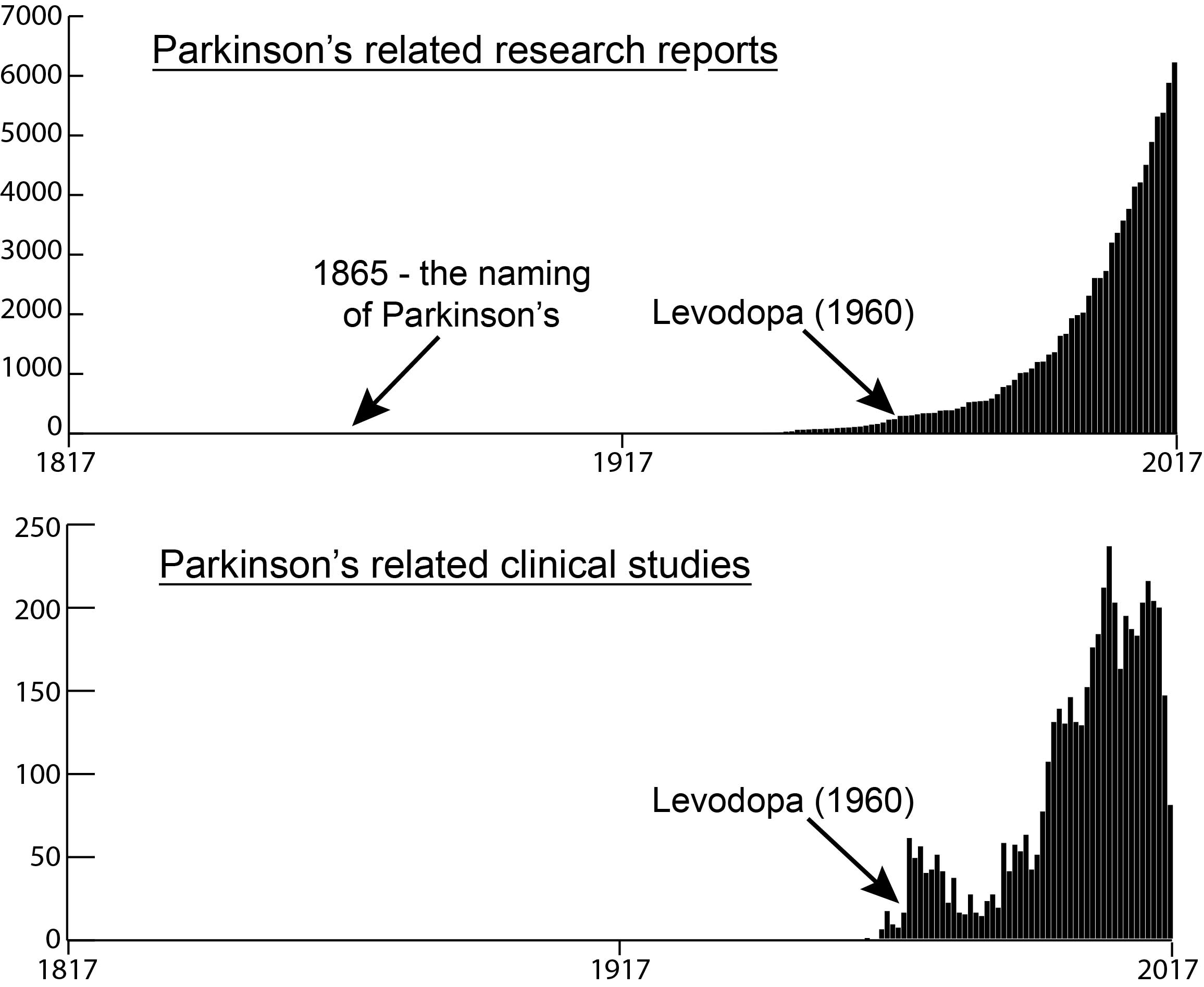
The number of research reports and clinical trial studies per year since 1817
As everyone in the Parkinson’s community is aware, in 2017 we were observing the 200th anniversary of the first description of the condition by James Parkinson (1817). But what a lot of people fail to appreciate is how little research was actually done on the condition during the first 180 years of that period.
The graphs above highlight the number of Parkinson’s-related research reports published (top graph) and the number of clinical study reports published (bottom graph) during each of the last 200 years (according to the online research search engine Pubmed – as determined by searching for the term “Parkinson’s“).
PLEASE NOTE, however, that of the approximately 97,000 “Parkinson’s“-related research reports published during the last 200 years, just under 74,000 of them have been published in the last 20 years.
That means that 3/4 of all the published research on Parkinson’s has been conducted in just the last 2 decades.
And a huge chunk of that (almost 10% – 7321 publications) has been done in 2017 only.
Last Monday, a SpaceX rocket lifted off from the Florida peninsular on route to the International Space Station.
On board that craft was an experiment that could have big implications for Parkinson’s disease. It involves a Parkinson’s-associated protein called Leucine-rich repeat kinase 2 (or LRRK2).
In today’s post, we will discuss why we needed to send this protein into orbit.
When you look up at the sky tonight – if you look for long enough – you may well see a bright little object hurtling across the sky (Click here to learn more about how to track the International Space Station). Know that inside that bright little object passing over you there is currently some Parkinson’s disease-related research being conducted.
What is the International Space Station?
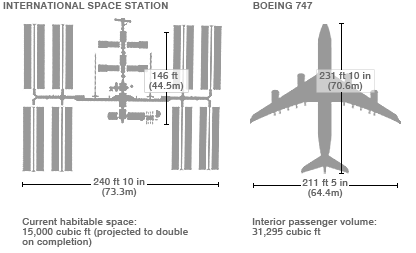
The International Space Station (or the ISS) is the largest human-made object that we have ever put into space. It is so big in fact that you can see it with the naked eye from Earth.
(How’s that for exciting viewing?)
The current space station is 73.3 metres (240 feet) long and 44.5 metres (146 feet) wide, weighing approximately 420 tonnes (924,740 lb), and it has been continuously occupied for 16 years and 289 days, making it the longest continuous human presence in low Earth orbit. The ISS travels at a speed of 7.67 km/second, maintains an altitude of between 330 and 435 km (205 and 270 mi), and completes 15.54 orbits per day (it has made over 102,000 orbits!).
The size of the the ISS compared to a Boeing Jumbo jet. Source: Reddit
First approved by President Ronald Reagan in 1984, it was not until November 1998 that the first components of the International space station were first launched into orbit. 36 shuttle flights were made to help build the station. The first crew members took up residence on the 2nd November 2000, and the station was completed in 2011. There is always 6 crew members on board – the current team are Expedition 52 – and it has been visited by 220 astronauts, cosmonauts and space tourists from 17 different nations since the project began.
The title of today’s post is written in jest – my job as a researcher scientist is to find a cure for Parkinson’s disease…which will ultimately make my job redundant! But all joking aside, today was a REALLY good day for the Parkinson’s community.
Last night (3rd August) at 23:30, a research report outlining the results of the Exenatide Phase II clinical trial for Parkinson’s disease was published on the Lancet website.
And the results of the study are good:while the motor symptoms of Parkinson’s disease subject taking the placebo drug proceeded to get worse over the study, the Exenatide treated individuals did not.
The study represents an important step forward for Parkinson’s disease research. In today’s post we will discuss what Exenatide is, what the results of the trial actually say, and where things go from here.
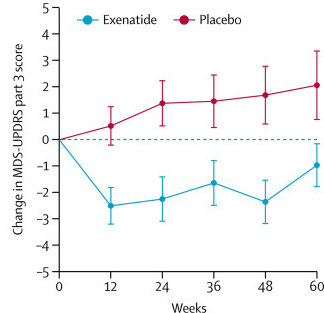
Last night, the results of the Phase II clinical trial of Exenatide in Parkinson’s disease were published on the Lancet website. In the study, 62 people with Parkinson’s disease (average time since diagnosis was approximately 6 years) were randomly assigned to one of two groups, Exenatide or placebo (32 and 30 people, respectively). The participants were given their treatment once per week for 48 weeks (in addition to their usual medication) and then followed for another 12-weeks without Exenatide (or placebo) in what is called a ‘washout period’. Neither the participants nor the researchers knew who was receiving which treatment.
At the trial was completed (60 weeks post baseline), the off-medication motor scores (as measured by MDS-UPDRS) had improved by 1·0 points in the Exenatide group and worsened by 2·1 points in the placebo group, providing a statistically significant result (p=0·0318). As you can see in the graph below, placebo group increased their UPDRS motor score over time (indicating a worsening of motor symptoms), while Exenatide group (the blue bar) demonstrated improvements (or a lowering of motor score).
Reduction in motor scores in Exenatide group. Source: Lancet
In October 2015, researchers from Georgetown University announced the results of a small clinical trial that got the Parkinson’s community very excited. The study involved a cancer drug called Nilotinib, and the results were rather spectacular.
What happened next, however, was a bizarre sequence of disagreements over exactly what should happen next and who should be taking the drug forward. This caused delays to subsequent clinical trials and confusion for the entire Parkinson’s community who were so keenly awaiting fresh news about the drug.
Earlier this year, Georgetown University announced their own follow up phase II clinical trial and this week a second phase II clinical trial funded by a group led by the Michael J Fox foundation was initiated.
In todays post we will look at what Nilotinib is, how it apparently works for Parkinson’s disease, what is planned with the new trial, and how it differs from the ongoing Georgetown Phase II trial.
This week the U.S. Food and Drug Administration (FDA) has given approval for a multi-centre, double-blind, randomised, placebo-controlled Phase IIa clinical trial to be conducted, testing the safety and tolerability of Nilotinib (Tasigna) in Parkinson’s disease.
This is exciting and welcomed news.
What is Nilotinib?
Nilotinib (pronounced ‘nil-ot-in-ib’ and also known by its brand name Tasigna) is a small-molecule tyrosine kinase inhibitor, that has been approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML).
What does any that mean?
Basically, it is the drug that is used to treat a type of blood cancer (leukemia) when the other drugs have failed. It was approved for treating this cancer by the FDA in 2007.
Dipraglurant is a mGluR5 negative allosteric modulator (don’t panic, it’s not as complicated as it sounds).
In today’s post, we’ll explain what all of that means and look at the science behind this new treatment.
An example of a person with dyskinesia. Source: JAMA Neurology
For anyone familiar with Parkinson’s disease, they will know that long term use of the treatment L-dopa can lead to two possible outcomes:
The treatment loses it’s impact, requiring ever higher doses to be administered
The appearance of dykinesias
Now, not everyone taking L-dopa will be affected by both of these outcomes, but people with young, onset Parkinson’s disease do seem to be at risk of developing L-dopa induced dykinesias.
What are Dyskinesias?
Dyskinesias (from Greek: dys – abnormal; and kinēsis – motion, movement) are simply a category of movement disorders that are characterised by involuntary muscle movements. And they are certainly not specific to Parkinson’s disease.
As we have suggested above, they are associated in Parkinson’s disease with long-term use of L-dopa.
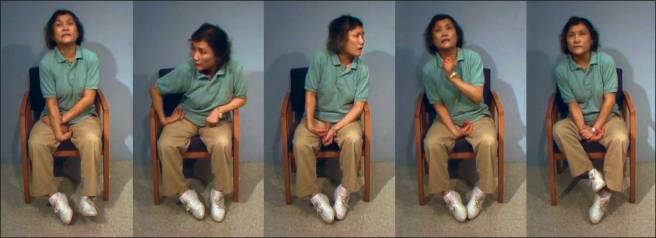
Below is a video of two legends: the late Tom Isaacs (who co-founded the Cure Parkinson’s Trust) and David Sangster (he founded www.1in20Parkinsons.org.uk). They were both diagnosed with Parkinson’s disease in their late 20’s. Tom, having lived with Parkinson’s for 20 years at the time of this video provides a good example of what dyskinesias look like:
As you can see, dyskinesias are a debilitating issue for anyone who suffers them.
How do dyskinesias develop in Parkinson’s disease?
Before being diagnosed and beginning a course of L-dopa, the locomotion parts of the brain in a person with Parkinson’s disease gradually becomes more and more inhibited. This increasing inhibition results in the slowness and difficulty in initiating movement that characterises this condition. A person with Parkinson’s may want to move, but they can’t.
They are akinetic (from Greek:a-,not,without; and kinēsis – motion).
Drawing of an akinetic individual with Parkinson’s disease, by Sir William Richard Gowers Source: Wikipedia
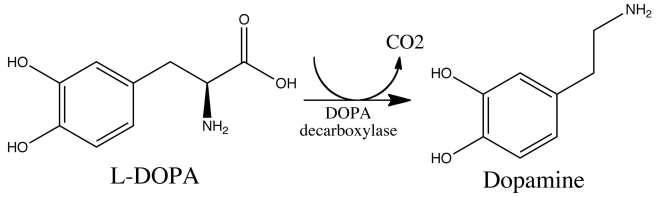
L-dopa tablets provide the brain with the precursor to the chemical dopamine. Dopamine producing cells are lost in Parkinson’s disease, so replacing the missing dopamine is one way to treat the motor features of the condition. Simply giving people pills of dopamine is a non-starter: dopamine is unstable, breaks down too quickly, and (strangely) has a very hard time getting into the brain. L-dopa, on the other hand, is very robust and has no problem getting into the brain.
Once inside the brain, L-dopa is quickly converted into dopamine. It is changed into dopamine by an enzyme called DOPA decarboxylase, and this change rapidly increases the levels of dopamine in the brain, allowing the locomotion parts of the brain to function more normally.
The chemical conversion of L-dopa to dopamine. Source: Nootrobox
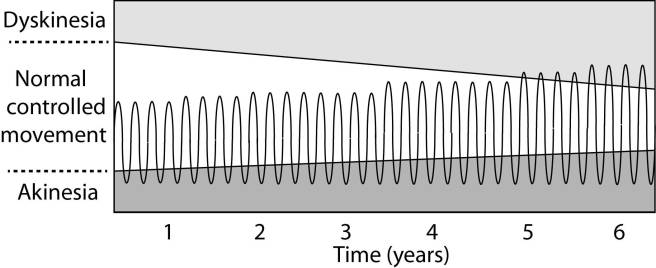
In understanding this process, it is important to appreciate that when an L-dopa tablet is consumed and L-dopa enters the brain, there is a rapid increase in the levels of dopamine. A ‘spike’ in the supply of dopamine, if you will, and this will last for the next few hours, before the dopamine is used up.
As the effects of the L-dopa tablet wear off, another tablet will be required. This use of multiple L-dopa pills across the day gives rise to a wave-like shape to the dopamine levels in the brain over the course of the day (see the figure below). The first pill in the morning will quickly lift the levels of dopamine enough that the individual will no longer feel akinetic. This will allow them to be able to function with normal controlled movement for several hours before the L-dopa begins to wear off. As the L-dopa wears off, the dopamine levels in the brain drop back towards levels that will leave the person feeling akinetic and at this point another L-dopa tablet is required.
After several years of L-dopa use, many people with Parkinson’s disease will experience a weaker response to each tablet. They will also find that they have more time during which they will be unable to move (exhibiting akinesia). This is simply the result of the progression of Parkinson’s disease – L-dopa treats the motor features of the disease but only hides/masks the fact that the disease is still progressing.
To combat this shorter response time, the dose of L-dopa is increased. This will result in increasing levels of dopamine in the brain (as illustrated by the higher wave form over time in the image below). It will take more L-dopa medication induced dopamine to lift the individual out of the akinetic state.
This increasing of L-dopa dosage, however, is often associated with the gradual development of abnormal involuntary movements that appear when the levels of L-dopa induced dopamine are the highest.
These are the dyskinesias.
Are there different types of dyskinesias?
Yes there are.
Dyskinesias have been broken down into many different subtypes, but the two main types of dyskinesia are:
Chorea – these are involuntary, irregular, purposeless, and unsustained movements. To an observer, Chorea will look like a very disorganised/uncoordinated attempt at dancing (hence the name, from the Greek word ‘χορεία’ which means ‘dance’). While the overall activity of the body can appear continuous, the individual movements are brief, infrequent and isolated. Chorea can cause problems with maintaining a sustained muscle contraction, which may result in affected people dropping things or even falling over.
Dystonia – these are sustained muscle contractions. They often occur at rest and can be either focal or generalized. Focal dystonias are involuntary contractions in a single body part, for example the upper facial area. Generalized dystonia, as the name suggests, are contraction affecting multiple body regions at the same time, typically the trunk, one or both legs, and another body part. The intensity of muscular movements in sufferers can fluctuate, and symptoms usually worsen during periods of fatigue or stress.
We have previously discussed the current treatment options for dyskinesias (click here to see that post).
Ok, so what clinical trials are Addex Therapeutics and the Michael J Fox Foundation preparing and why?
They are preparing to take a drug called Dipraglurant through phase III testing for L-dopa inducing dyskinesias in Parkinson’s disease. Dipraglurant is a mGluR5 negative allosteric modulator.
And yes, I know what you are going to ask next: what does any of that mean?
Ok, so mGluR5 (or Metabotropic glutamate receptor 5) is a G protein-coupled receptor. This is a structure that sits in the skin of a cell (the cell membrane), with one part exposed to the outside world – waiting for a chemical to bind to it – while another part is inside the cell, ready to act when the outside part is activated. The outside part of the structure is called the receptor.
Metabotropic receptors are a type of receptor that is indirectly linked with channels in cell membrane. These channels open and close, allowing specific elements to enter the cell. When a chemical (or agonist) binds to the receptor and it becomes activated, the part of the structure inside the cell will send a signal to the channel via a messenger (called a G-protein).
The chemical that binds to mGluR5 is the neurotransmitter glutamate.
But what about the “negative allosteric modulator” part of ‘mGluR5 negative allosteric modulator’
Good question.
This is the key part of this new approach. Allosteric modulators are a new class of orally available small molecule therapeutic agents. Traditionally, most marketed drugs bind directly to the same part of receptors that the body’s own natural occurring proteins attach to. But this means that those drugs are competing with those endogenous proteins, and this can limit the potential effect of the drug.
Allosteric modulators get around this problem by binding to a different parts of the receptor. And instead of simply turning on or off the receptor, allosteric modulators can either turn up the volume of the signal being sent by the receptor or decrease the signals. This means that when the body’s naturally occurring protein binds in the receptor, allosteric modulators can either amplify the effect or reduce it depending on which type of allosteric modulators is being administered.
There are two different types of allosteric modulators: positive and negative. And as the label suggests, positive allosteric modulators (or PAMs) increase the signal from the receptor while negative allosteric modulators (or NAMs) reduce the signal.
So Dipraglurant turns down the volume of the signal from the mGluR5 receptor?
Exactly.
By turning down the volume of the glutamate receptor mGluR5, researchers believe that we can reduce the severity of dyskinesias.
But hang on a second. Why are we looking at glutamate in dyskinesias? Isn’t dopamine the chemical of interest in Parkinson’s disease?
So almost 10 years ago, some researchers noticed something interesting in the brains of Parkinsonian monkeys that had developed dyskinesias:
Title: mGluR5 metabotropic glutamate receptors and dyskinesias in MPTP monkeys. Authors: Samadi P, Grégoire L, Morissette M, Calon F, Hadj Tahar A, Dridi M, Belanger N, Meltzer LT, Bédard PJ, Di Paolo T. Journal: Neurobiol Aging. 2008 Jul;29(7):1040-51. PMID:17353071
The researchers conducting this study induced Parkinson’s disease in monkeys using a neurotoxin called MPTP, and they then treated the monkeys with L-dopa until they began to develop dyskinesias. At this point when they looked in the brains of these monkeys, the researchers noticed a significant increase in the levels of mGluR5, which was associated with the dyskinesias. This finding led the researchers to speculate that reducing mGluR5 levels might reduce dyskinesias.
And it did!
Subsequent preclinical research indicated that targeting mGluR5 might be useful in treating dyskinesias, especially with negative allosteric modulators:
Title: The mGluR5 negative allosteric modulator dipraglurant reduces dyskinesia in the MPTP macaque model Authors: Bezard E, Pioli EY, Li Q, Girard F, Mutel V, Keywood C, Tison F, Rascol O, Poli SM. Journal: Mov Disord. 2014 Jul;29(8):1074-9. PMID:24865335
In this study, the researchers tested the efficacy of dipraglurant in Parkinsonian primates that had developed L-dopa induced dyskinesias. They tested three different doses of the drug (3, 10, and 30 mg/kg).
Dipraglurant significantly reduced dyskinesias in the monkeys, with best effect being reached using the 30 mg/kg dose. Importantly, the dipraglurant treatment had no impact on the efficacy of L-dopa which was still being used to treat the monkeys Parkinson’s features.
This research lead to a clinical trials in man, and last year Addex Therapeutics published the results of their phase IIa clinical trial of Dipraglurant (also called ADX-48621):
Title: A Phase 2A Trial of the Novel mGluR5-Negative Allosteric Modulator Dipraglurant for Levodopa-Induced Dyskinesia in Parkinson’s Disease. Authors: Tison F, Keywood C, Wakefield M, Durif F, Corvol JC, Eggert K, Lew M, Isaacson S, Bezard E, Poli SM, Goetz CG, Trenkwalder C, Rascol O. Journal: Mov Disord. 2016 Sep;31(9):1373-80. PMID:27214664
The Phase IIa double-blind, placebo-controlled, randomised trial was a dose escalation study, conducted in 76 patients with Parkinson’s disease L-dopa-induced dyskinesia – 52 subjects were given dipraglurant and 24 received a placebo treatment. The dose escalation assessment of dipraglurant started at 50 mg once daily to 100 mg 3 times daily. The study was conducted over 4 weeks.
The investigators found that dipraglurant significantly reduced the dyskinesias on both day 1 of the study and on day 14, and this treatment did not result in any worsening of the Parkinsonian features. And remember that this was a double blind study, so both the investigators and the participants had no idea which treatment was being given to each subject. Thus little bias can influence the outcome, indicating that dipraglurant really is having a beneficial effect on dyskinesias.
The company suggested that dipraglurant’s efficacy in reducing L-dopa-induced dyskinesia warrants further investigations in a larger number of patients. And this is what the company is now doing with the help of the Michael J. Fox Foundation (MJFF). In addition, dipraglurant’s potential benefits on dystonia are also going to be investigated with support from the Dystonia Medical Research Foundation (DMRF).
And the really encouraging aspect of this research is that Addex Therapeutics are not the only research group achieving significant beneficial results for dykinesias using this treatment approach (click here to read about other NAM-based clinical studies for dyskinesias).
Fingers crossed for more positive results here.
What happens next?
L-dopa induced dyskinesias can be one of the most debilitating aspects of living with Parkinson’s disease, particularly for the early-onset forms of the condition. A great deal of research is being conducted in order to alleviate these complications, and we are now starting to see positive clinical results starting to flow from that research.
These results are using new type of therapeutic drug that are designed to increase or decrease the level of a signal occurring in a cell without interfering with the normal functioning of the chemicals controlling the activation of that signal.
This is really impressive biology.
The banner for today’s post was sourced from Steam
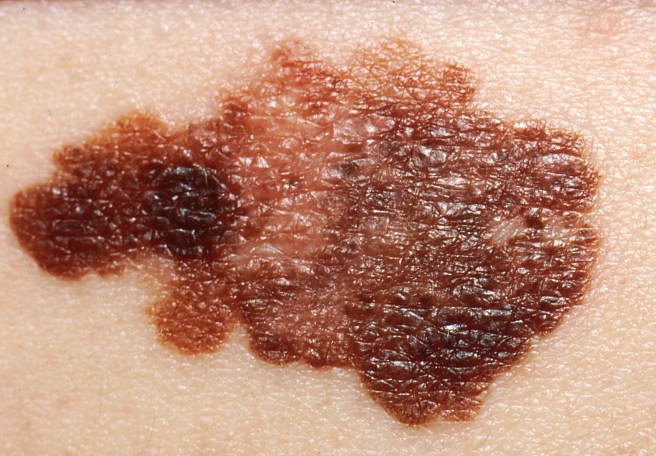
A build up of a protein called alpha synuclein inside neurons is one of the characteristic feature of the Parkinsonian brain. This protein is believed to be partly responsible for the loss of dopamine neurons in this condition.
A similar build up of alpha synuclein is also seen in the deadly skin cancer, Melanoma… but those cells don’t die (?!?)… in fact, they just keep on dividing.
Why is there this critical difference?
In today’s post we look at an interesting new study that may have solved this mystery.
The truly baffling detail in this story, however, is that this relationship is reciprocal – if you have melanoma you are almost 3 times more likely to develop Parkinson’s disease than someone without melanoma (Source: Baade et al 2007; Gao et al 2009).
What is melanoma exactly?
Melanoma is a type of skin cancer.
It develops from the pigment-containing cells known as melanocytes. Melanocytes are melanin-producing cells located in the bottom layer (the stratum basale) of the skin’s outer layer (or epidermis).
The location of melanocytes in the skin. Source: Wikipedia
Melanocytes produce melanin, which is a pigment found in the skin, eyes, and hair. It is also found in the brain in certain types of cells, such as dopamine neurons (where it is referred to as neuromelanin).
Neuromelanin (brown) in dopamine neurons. Source: Schatz
Melanomas are usually caused by DNA damage resulting from exposure to ultraviolet radiation. Ultraviolet radiation from tanning beds increases the risk of melanoma (Source), as does excessive air travel (Source), or simply spending to much time sun bathing.
Approximately 2.2% of men and women will be diagnosed with melanoma at some point during their lives (Source).In women, melanomas most commonly occur on the legs, while in men they are most common on the back. Melanoma makes up 5% of all cancers (Source).
Generally, melanomas is one of the safer cancers, as it can usually be detected early by visual inspection. This cancer is made dangerous, however, by its ability to metastasise (or spread to other organs in the body).
Are there any genetic associations between Parkinson’s disease and melanoma?
No.
When the common genetics mutations that increase the risk of both conditions were previously analysed, it was apparent that none of the known Parkinson’s mutations make someone more susceptible to melanoma, and likewise none of the melanoma-associated genetic mutations make a person vulnerable to Parkinson’s disease (Meng et al 2012;Dong et al 2014; Elincx-Benizri et al 2014).
In fact, researchers have only found very weak genetic connections between two conditions (Click here to read our previous post on this). It’s a real mystery.
Are there any other connections between Parkinson’s disease and melanoma?
Yes.
Another shared feature of both Parkinson’s disease and melanoma is the build up of a protein called alpha synuclein. Alpha synuclein is believed to be one of the villains in Parkinson’s disease – building up inside a cell, becoming toxic, and eventually killing that cell.
But recently researchers noticed that melanoma also has a build up of alpha synuclein, but those cells don’t die:
Title: Parkinson’s disease-related protein, alpha-synuclein, in malignant melanoma Authors: Matsuo Y, Kamitani T. Journal: PLoS One. 2010 May 5;5(5):e10481. PMID:20463956 (This article is OPEN ACCESS if you would like to read it)
In this study, researchers from Japan found that alpha synuclein was detected in 86% of the primary and 85% of the metastatic melanoma. Understand that the protein is not detectable in the non-melanoma cancer cells.
So what is it doing in melanoma cells?
Recently, researchers from Germany believe that they have found the answer to this question:
Title: Treatment with diphenyl-pyrazole compound anle138b/c reveals that α-synuclein protects melanoma cells from autophagic cell death Authors: Turriani E, Lázaro DF, Ryazanov S, Leonov A, Giese A, Schön M, Schön MP, Griesinger C, Outeiro TF, Arndt-Jovin DJ, Becker D Journal: Proc Natl Acad Sci U S A. 2017 Jun 5. pii: 201700200. doi: 10.1073/pnas.1700200114 PMID:28584093
In their study, the German researchers looked at levels of alpha synuclein in melanoma cells. They took the melanoma cells that produced the most alpha synuclein and treated those cells with a chemical that inhibits the toxic form of alpha synuclein (which results from the accumulation of the protein).
What they observed next was fascinating: the cell morphology (or physically) changed, leading to massive melanoma cell death. The investigators found that this cell death was caused by instability of mitochondria and a major dysfunction in the autophagy process.
Mitochondria, you may recall, are the power house of each cell. They keep the lights on. Without them, the lights go out and the cell dies.
Mitochondria and their location in the cell. Source: NCBI
Autophagy is the garbage disposal/recycling process within each cell, which is an absolutely essential function. Without autophagy, old proteins and mitochondria will pile up making the cell sick and eventually it dies. Through the process of autophagy, the cell can break down the old protein, clearing the way for fresh new proteins to do their job.
Waste material inside a cell is collected in membranes that form sacs (called vesicles). These vesicles then bind to another sac (called a lysosome) which contains enzymes that will breakdown and degrade the waste material. The degraded waste material can then be recycled or disposed of by spitting it out of the cell.
What the German research have found is that the high levels of alpha synuclein keep the mitochondria stable and the autophagy process working at a level that helps to keeps the cancer cell alive.
Next, they replicated this cell culture research in mice with melanoma tumors. When the mice were treated with the chemical that inhibits the toxic form of alpha synuclein, the cancer cancer became malformed and the autophagy process was blocked.
The researchers concluded that “alpha synuclein, which in PD exerts severe toxic functions, promotes and thereby is highly beneficial to the survival of melanoma in its advanced stages”.
So what does all of this mean for Parkinson’s disease?
Well, this is where the story gets really interesting.
You may be pleased to know that the chemical (called Anle138b) which was used to inhibit the toxic form of alpha synuclein in the melanoma cells, also works in models of Parkinson’s disease:
Title: Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Authors: Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, Geissen M, Eiden M, Leidel F, Hirschberger T, Deeg AA, Krauth JJ, Zinth W, Tavan P, Pilger J, Zweckstetter M, Frank T, Bähr M, Weishaupt JH, Uhr M, Urlaub H, Teichmann U, Samwer M, Bötzel K, Groschup M, Kretzschmar H, Griesinger C, Giese A. Journal: Acta Neuropathol. 2013 Jun;125(6):795-813 PMID:23604588 (This article is OPEN ACCESS if you would like to read it)
In this first study the researchers discovered Anle138b by conducted a large screening study to identify for molecules that could inhibit the toxic form of alpha synuclein.
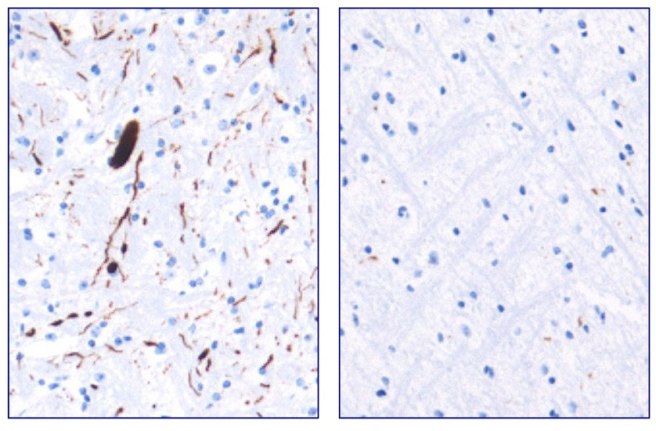
They next tested Anle138b in both cell culture and rodent models of Parkinson’s disease and found it to be neuroprotective and very good at inhibiting the toxic form of alpha synuclein. And the treatment looks to be very effective. In the image below you can see dark staining of toxic alpha synuclein in the left panel from the brain of an untreated mouse, but very little staining in the right panel from an Anle138b treated mouse.
Toxic form of alpha synuclein (dark staining). Source: Max-Planck
Importantly, Anle138b does not interfere with normal behaviour of alpha synuclein in the mice (such as production of the protein, correct functioning, and eventual degradation/disposal of the protein), but it does act as an inhibitor of alpha synuclein clustering or aggregation (the toxic form of the protein). In addition, the investigators found no toxic effects of Anle138b in any of their experiments even after long-term high-dose treatment (more than one year).
And in a follow up study, the drug was effective even if it was given after the disease model had started:
Title: The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset Authors: Levin J, Schmidt F, Boehm C, Prix C, Bötzel K, Ryazanov S, Leonov A, Griesinger C, Giese A. Journal: Acta Neuropathol. 2014 May;127(5):779-80. PMID:24615514 (This article is OPEN ACCESS if you would like to read it)
During the first study, the researchers had started Anle138b treatment in the mouse model of Parkinson’s disease at a very young age. In this study, however, the investigators began treatment only as the symptoms were starting to show, and Anle138b was found to significantly improve the overall survival of the mice.
One particularly interesting aspect of Anle138b function in the brain is that it does not appear to change the level of the autophagy suggesting that the biological effects of treatment with Anle138b is cell-type–specific (Click here to read more about this). In cancer cells, it is having a different effect to that in brain cells. These differences in effect may also relate to disease conditions though, as Anle138b was not neuroprotective in a mouse model of Multiple System Atrophy (MSA; Click here to read more about this).
Is Anle138b being tested in the clinic?
Not yet.
Ludwig-Maximilians-Universität München and the Max Planck Institute for Biophysical Chemistry (Göttingen) have spun off a company called MODAG GmbH that is looking to advance Anle138b to the clinic (Click here for the press release). The Michael J Fox Foundation are helping to fund more preclinical development of this treatment (Click here to read more about this).
We will be watching their progress with interest.
What does it all mean?
Summing up: There are many mysteries surrounding Parkinson’s disease, but some researchers from Germany may have just solved one of them and at the same time developed a potentially useful new treatment.
They have discovered that the Parkinson’s associated protein, alpha synuclein, which is produced in large amounts in the skin cancer melanoma, is actually playing an important role in keeping those cancer cells alive. By finding a molecule that can block the build up of alpha synuclein, they have not only found a treatment for melanoma, but also potentially one for Parkinson’s disease.
And given that both diseases are closely associated, this could be seen as a great step forward. Two birds with one stone as the saying goes.
The banner for today’s post was sourced from Wikipedia

 Source: DenisonMag
Source: DenisonMag  The foundation has just one mission: “to actively pursue and invest in cutting edge research with the goal of discovering new therapies for the treatment of Parkinson’s Disease in GBA mutation carriers”
The foundation has just one mission: “to actively pursue and invest in cutting edge research with the goal of discovering new therapies for the treatment of Parkinson’s Disease in GBA mutation carriers”