
|
This week the ‘Michael J. Fox Foundation for Parkinson’s Research’ and ‘The Silverstein Foundation for Parkinson’s with GBA’ announced that they are collaboratively awarding nearly US$3 million in research grants to fund studies investigating an enzyme called beta glucocerebrosidase (or GCase). Why is this enzyme important to Parkinson’s? In today’s post, we will discuss what GCase does, how it is associated with Parkinson’s, and review what some of these projects will be exploring. |
 Source: DenisonMag
Source: DenisonMag
This is Jonathan Silverstein.
He is a General Partner of Global Private Equity at OrbiMed – the world’s largest fully dedicated healthcare fund manager. During his time at OrbiMed, the company has invested in healthcare companies that have been involved with over 60 FDA approved products.
In February 2017 – at just 49 years of age – Jonathan was diagnosed with Parkinson’s.
Rather than simply accepting this diagnosis, however, Mr Silverstein decided to apply the skills that he has built over a long and successful career in funding biotech technology, and in March 2017, he and his wife, Natalie, set up the Silverstein Foundation for Parkinson’s with GBA.
 The foundation has just one mission: “to actively pursue and invest in cutting edge research with the goal of discovering new therapies for the treatment of Parkinson’s Disease in GBA mutation carriers”
The foundation has just one mission: “to actively pursue and invest in cutting edge research with the goal of discovering new therapies for the treatment of Parkinson’s Disease in GBA mutation carriers”
And it seeks to address this by achieving three goals:
- to find a way to halt the progression of Parkinson’s with GBA.
- to identify regenerative approaches to replace the damaged/lost cells
- to find preventative measures
This week, the Silverstein foundation and the Michael J. Fox Foundation for Parkinson’s Research made a big anoouncement.

The two organisations announced nearly US$3 million in grants to fund studies investigating an enzyme called glucocerebrosidase beta acid (or GCase).
And what exactly is glucocerebrosidase?
Glucocerebrosidase/GCase is an enzyme that helps with the digestion and recycling of various proteins (particularly glucocerebrosides) inside cells. The enzyme is located inside lysosomes.
What are lysosomes?
Lysosomes are the small bags of digestive enzymes inside cells.
How do these lysosomes work?
On a continual basis, small parts of the external layer of the cell membrane is brought inside the cell. This is a process called endocytosis. It occurs when the cell consumes resources from the outside world in order to find what it needs to function and survive. As a section of cell membrane is brought into the cell, it forms a vesicle (which is a term used to refer to small spherical bags of stuff inside cells). Given the process by which that vesicles was formed, it is referred to as an endosome (sometimes it is also called a vacuole).
 Source: Socratic
Source: Socratic
Once the endosome is inside the cell and detached from the rest of the membrane, it will bind to another vesicle – this one being the lysosome. And as I mentioned above, lysosome is a small bag that is full of digestive enzymes, which help to break down the contents of the endosome.

How lysosomes work. Source: Prezi
The lysosome will fuse with the endosome/vacuole and the enzymes from the lysosome will mix with the material in the vacuole and digest it (or it break down into more manageable components).
This enzymatic process works in a very similar fashion to the commercial products that you use for washing your clothes.
 Enzymatic degradation. Source: Samvirke
Enzymatic degradation. Source: Samvirke
The reagents that you put into the washing machine with your clothes contain a multitude of enzymes, which help to break down the dirty, bacteria, flakes of skin, etc that cling to your clothes. Each enzyme breaks down a particular protein, fat or such like. And this is very similar to the collection of enzymes in the lysosome. All of them are needed to break down all of the contents of the endosome.
And if one of those enzymes – such as glucocerebrosidase – is faulty (due to a genetic mutation), then the enzymatic process is disrupted, which could result in the build up of un-degraded material over time.
Ok, but what does this have to do with Parkinson’s?
The enzyme Glucocerebrosidase is produced by cells using instructions provided by the GBA gene (a gene is a section of DNA that provides the instructions for making a particular protein).
Genetic variations in the GBA gene are associated with two medical conditions:
- Parkinson’s
- Gaucher disease.
What is Gaucher disease?
Gaucher (pronounced “go-shay”) disease is a rare inherited genetic disorder characterised by the build up in cells of a fatty chemical called glucocerebroside. Because the body cannot break down this chemical, swollen fat-laden cells build up in certain areas of the body, such as the spleen, liver and bone marrow. These cells are referred to as ‘Gaucher cells’.
 Swollen Gaucher cells (circled in red). Source: Imagebank
Swollen Gaucher cells (circled in red). Source: Imagebank
The disorder results from the deficiency of the enzyme glucocerebrosidase, which usually breaks down glucocerebroside. The incidence of Gaucher disease is about one in every 40,000 live births (Source), and the condition manifests itself in several way, from reduced bone density to swollen liver and spleen:
 The signs of Gaucher disease. Source: Rare2aware
The signs of Gaucher disease. Source: Rare2aware
What happens to the cells in Gaucher disease?
Macrophage are one type of cell that is particularly affected in Gaucher disease. They are a type of blood cell that is responsible for detecting, engulfing and destroying dangerous pathogens and apoptotic cells. Below is a schematic of a macrophage, consuming orange pathogens (left), digesting them, and releasing the waste (on the right):
 A schematic of a macrophage. Source: Meducator
A schematic of a macrophage. Source: Meducator
Macrophage travel the body, swallowing anything that they don’t like the look of. In order to break down everything it swallows, a macrophage must have a full complement of digestive enzymes. But – as you can see in the image below – without glucocerebrosidase, the macrophage has trouble digesting fatty chemicals like glucocerebrosides and the lysosomes start to accumulate in the cell, causing the cell to swell up.
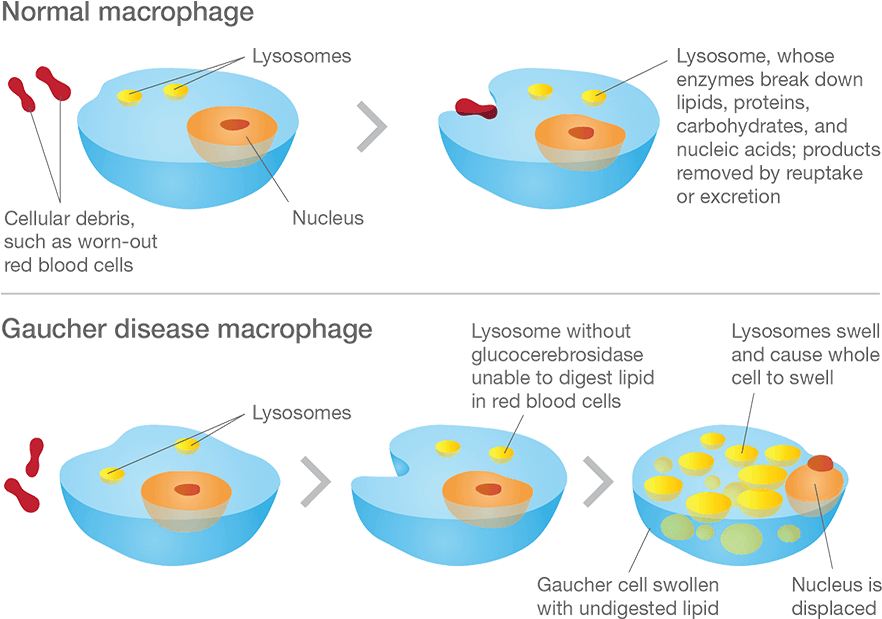 Gaucher disease. Source: Gaucherawareness
Gaucher disease. Source: Gaucherawareness
Are there different types of Gaucher disease?
There are three types of Gaucher disease, and they are all caused by genetic mutations in the GBA gene:
- Type I – (also called the “non-neuropathic” type) this is the most common; having said that it mainly occurs in Ashkenazi Jews (x100 more than the general population). The median age at diagnosis is 28 years of age, and life expectancy is only mildly decreased. As the “non-neuropathic” label suggests, there are no neurological symptoms.
- Type II – is characterised by neurological problems in small children. The glucocerebrosidase enzyme is barely present in the lysosomes. Prognosis is poor (death before the age of three).
- Type III – (the Swedish variety) occurs in people from the Norrbotten region in Sweden. This group develops the disease somewhat later, but most most do not survive their 30th birthday.
There are three subtypes of Gaucher disease, which are characterised by the presence or absence of neurological issues, and by the rate of disease progression and severity:
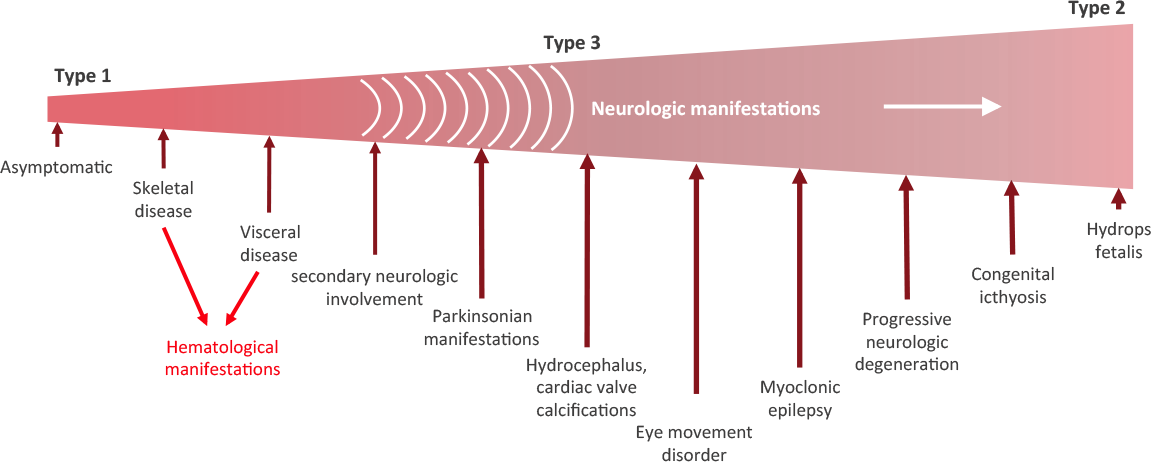 Stages of Gaucher disease. Source: Gaucherawareness
Stages of Gaucher disease. Source: Gaucherawareness
You said Gaucher disease is inherited. So if one of my parents has Gaucher disease will it affect me?
Gaucher disease is an inherited genetic disorder, caused by mutations in the GBA gene. Humans normally have two copies of the GBA gene. If one copy of the GBA gene is faulty due to a genetic mutation, the person will not develop Gaucher’s disease, because the one remaining functional gene will be able to produce enough of the glucocerebrosidase enzyme.
Gaucher disease is considered an autosomal recessive disorder. This means that two copies of an abnormal gene must be present in the individual in order for the disease to develop. A person with just one faulty gene will not get sick, but they will be a carrier. To develop Gaucher disease, you need to have two genetic mutations in the GBA gene – one from your mother and one from your father.
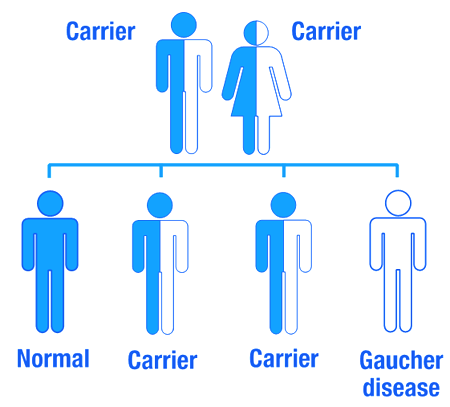 An autosomal recessive disorder. Source: Myhealthyfeeling
An autosomal recessive disorder. Source: Myhealthyfeeling
So even if one of your parents has been diagnosed with Gaucher disease, you will not necessarily develop it if the other parent does not have a mutation in their GBA gene.
Ok. All of this is interesting, but how is Gaucher disease associated with Parkinson’s?
In the 1990s, physicians began to notice patients with both Gaucher and Parkinson’s. An example of this was a report published in 1996 that described six people with Gaucher disease who also exhibited an early-onset, severe form of Parkinson’s with cognitive decline:
 Title: Occurrence of Parkinson’s syndrome in type I Gaucher disease.
Title: Occurrence of Parkinson’s syndrome in type I Gaucher disease.
Authors: Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A.
Journal: QJM. 1996 Sep;89(9):691-4.
PMID: 8917744 (This article is OPEN ACCESS if you would like to read it)
In this study, the Israeli researchers report on 6 people with Type I Gaucher disease (which up until that point had not been considered neuronopathic). All six of the subjects also exhibited the hallmark of a rather severe form of Parkinson’s, which made its appearance in the 4th to 6th decade of life and displayed an aggressive progression and was largely unresponsive to conventional anti-Parkinson therapy (such as L-dopa).
These initial reports were followed by many additional studies which eventually started pointing towards the GBA gene as the likely risk factor for this form of Parkinson’s, including this study:
 Title: Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews.
Title: Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews.
Authors: Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R.
Journal: N Engl J Med. 2004 Nov 4;351(19):1972-7.
PMID: 15525722 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators examined 99 individuals with idiopathic Parkinson’s from an Ashkenazi Jewish background. Thirty-one of them (31.3%) had one or two mutations in their GBA gene. And of all the individuals with Parkinson’s, the subjects who were carriers of GBA mutations were younger than those who were not carriers (mean age at onset being 60 years vs. 64 years).
And this result is similar to what has been seen in larger follow up studies (Click here for an example).
So genetic variations in the GBA gene are associated with Parkinson’s?
Yes. It is one of the main genetic risk factors for the condition.
It is now believed that approximately 5%–8% of people with Parkinson’s have a genetic mutation in the GBA gene (Click here and here to read more about this). According to the Michael J Fox foundation webpage on GBA “up to 10 percent of people with PD in the United States carry” a genetic variation in the GBA gene.
What do we know about the GBA gene?
GBA is a very large gene and there are numerous genetic variants spread across its length. The most common mutations are located in positions N370S and L444P. These mutations cause a reduction in the enzymatic activity of the glucocerebrosidase enzyme.
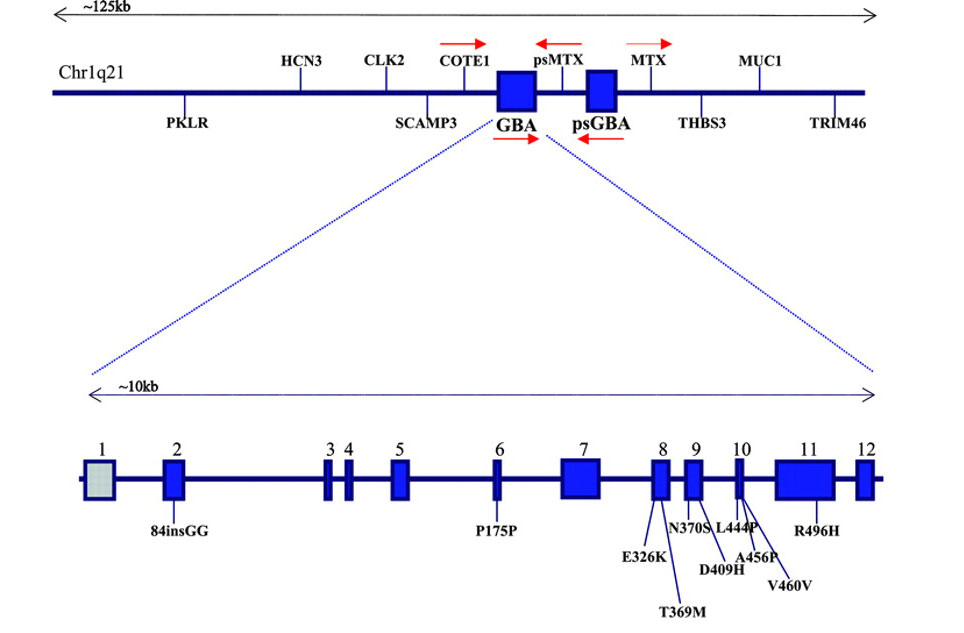 The GBA gene and mutations associated with Parkinson’s. Source: Neurology
The GBA gene and mutations associated with Parkinson’s. Source: Neurology
And as I mentioned above generally, people with GBA-associated Parkinson’s will exhibit more progressive symptoms than people without a GBA mutation, and this has recently been confirmed in a large clinical observation study:
 Title: Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study.
Title: Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study.
Authors: Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, Bajaj N, Barker RA, Burn DJ, Foltynie T, Hardy J, Wood NW, Ben-Shlomo Y, Williams NW, Grosset DG, Morris HR; PRoBaND clinical consortium.
Journal: J Neurol Neurosurg Psychiatry. 2018 [Epub ahead of print]
PMID: 29378790 (This article is OPEN ACCESS if you would like to read it)
In this multi-research centre study, the investigators studied 1893 people with Parkinson’s. Of these 171 (9.0%) had one copy of a known GBA mutation, of whom 121 (6.2%) had a genetic variant known to be associated with Parkinson’s but not causing Gaucher’s disease, and 50 (2.7%) had a genetic variant linked to both Parkinson’s and Gaucher’s. Another 28 (1.5%) carried genetic variants of unknown significance in the GBA gene. The most common GBA mutations associated with Parkinson’s in this study were ‘E362K‘, ‘T369M’ , and ‘L444P’.
Individuals with Parkinson’s associated GBA mutations were on average diagnosed 5 years earlier compared with non-carriers. They were also more likely to have postural instability and gait difficulties compared with non-carriers. In addition, they had more progressive forms of Parkinson’s (as determined by more advanced Hoehn and Yahr staging – after adjustment for age – compared with non-carriers).
No differences was observed in cognitive function between GBA mutation carriers and non-carriers. Cognitive impairment/dementia have been reported in other studies at a later stages of the condition (Click here to read more about this). This observation led the researchers to conclude that “this offers an important window of opportunity for potential disease-modifying therapy that may protect against the development of dementia”.
This is really bad, right? What if I have a GBA genetic mutation?
It is important for readers to understand that our understanding of the genetics of Parkinson’s is basic at best and that this aggressive pattern of disease progression in GBA-associated Parkinson’s is not always the case.
In addition, 2-3% of the general population will have a Parkinson’s associated GBA mutation in their DNA, but they never go on to present any of the features of Parkinson’s. And then there are cases of identical twins who both have a GBA mutations, but only one of them has developed Parkinson’s (Click here to read more about this).
Thus, the genetics of Parkinson’s is still very complex, and just because a person has a GBA mutation, it does not necessarily mean that they will go on to develop Parkinson’s or necessarily present the aggressive form of the condition.
More research is required to increase our understanding of all aspects of GCase and GBA.
And that brings us back to the announcement this week from the Silverstein foundation and the Michael J Fox Foundation.
Ok, so what did the Silverstein and Michael J Fox Foundations announce this week?
The two organisations announced almost US$3 million in awarded research grants to focus on GBA-associated Parkinson’s.
In the press release, Silverstein Foundation Founder Jonathan Silverstein said, “We are very pleased with the collaboration with The Michael J. Fox Foundation and feel confident that the projects chosen will significantly add to the library of knowledge around GBA and propel new treatments for people living with Parkinson’s and, perhaps, individuals at risk for the disease.”
And it is an extremely interesting line up of studies.
- Dr Tim Ahfeldt (Icahn School of Medicine at Mount Sinai) will use CRISPR gene-editing technology to modulate the levels of GBA and other associated genes in an effort to identify what other genetic risk factors could be at play in GBA-associated Parkinson’s (Click here to read more about CRISPR gene editing)
- Dr Justin Martin O’Sullivan (University of Auckland in New Zealand – YAY!) will use computer software to identify genes controlled by the DNA switches in the GBA gene and then investigate how these switches affect cells and may contribute to Parkinson’s.
- Dr Ricardo Feldman (University of Maryland) will use induced pluripotent stem (IPS) cells to examine the cellular effects of GBA mutations, and will then test if reversing some of those effects can actually protect dopamine neurons.
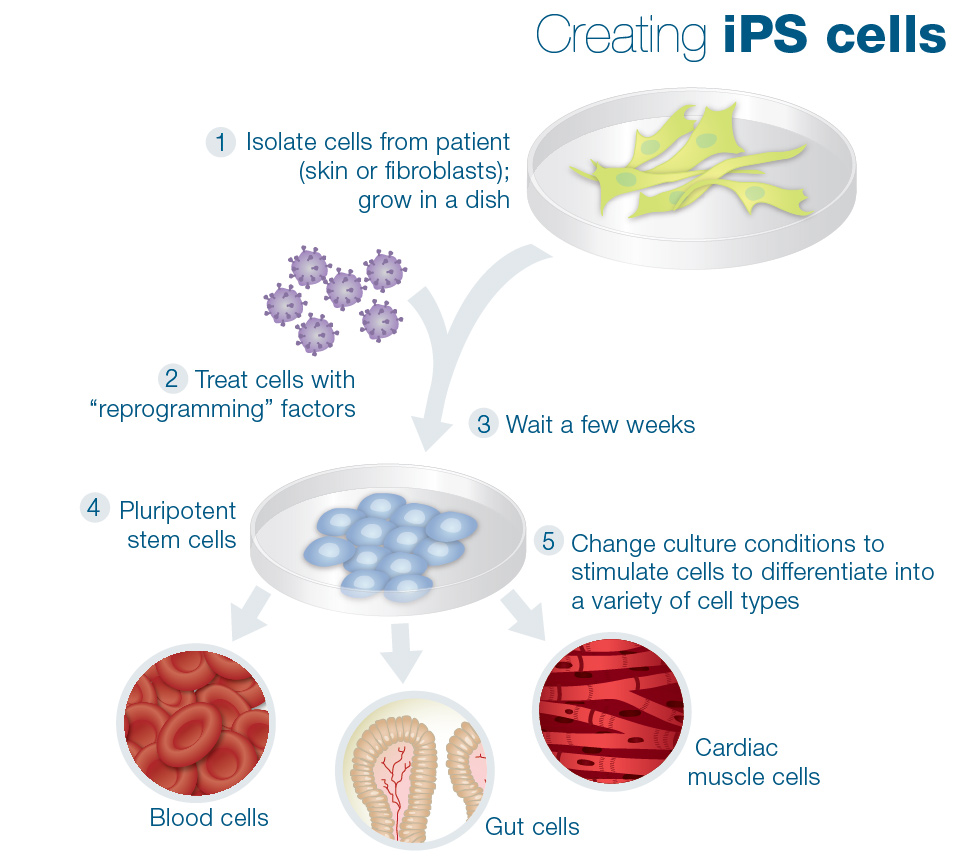 Making IPS cells. Source: learn.genetics
Making IPS cells. Source: learn.genetics
- Prof Anthony Futerman (Weizmann Institute of Science in Israel) will apply advanced RNA sequencing and proteomics analysis to postmortem brain tissue samples from people with idiopathic Parkinson’s, GBA-associated Parkinson’s, and control volunteers to identify GBA-specific pathology.
- Dr Manoj Kumar Pandey (Cincinnati Children’s Hospital Medical Center) is studying how GBA genetic mutations may lead to increased levels of inflammation.
- Prof Michel Desjardins (Université de Montréal in Canada) will study the role of GCase in autoimmune mechanisms after observing that GCase expression in immune cells influences this response (ooh, now this is interesting!).
- Dr Emily M. Rocha and Prof Tim Greenamyre (University of Pittsburgh) will focus on connecting deficits in GCase with another leading genetic target: LRRK2 (This work could explore the potential of LRRK2 inhibitors which are currently in clinical trials for Parkinson’s – Click here to read more about LRRK2 and those clinical trials).
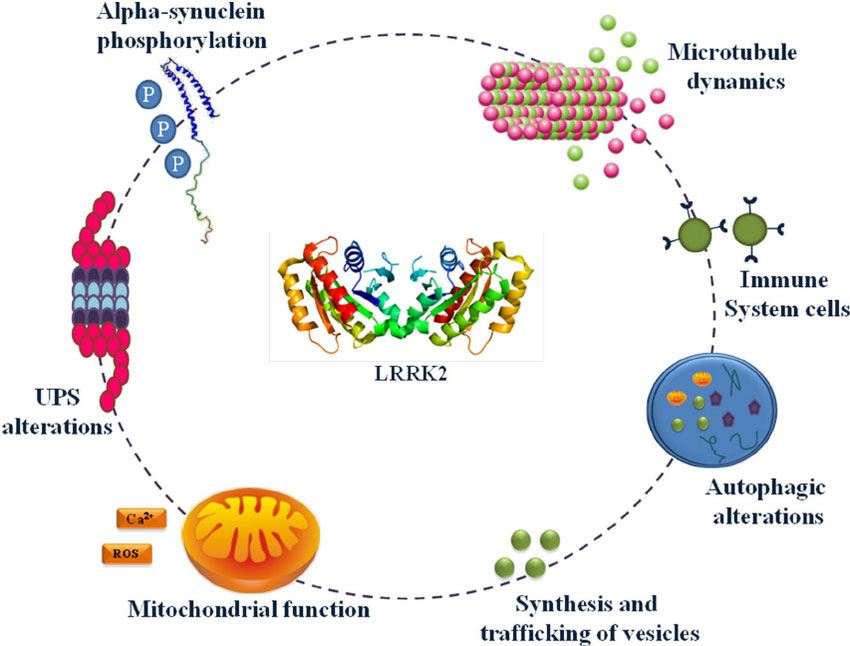 The many jobs of LRRK2. Source: Researchgate
The many jobs of LRRK2. Source: Researchgate
- Dr Aarnoud Cornelis van der Spoel (Dalhousie University in Canada) will investigate how sphingolipids are impacted by GBA mutations (Click here to read more about sphingolipids).
- Dr Peter Vangheluwe (KU Leuven research university in Belgium) will be looking at a transporter of the lipid glucosylceramide, which accumulates in response to GBA mutations.
- Friederike Zunke (University of Keil in Germany) will be searching for the site of interaction between GCase and the protein LIMP-2, which is known to transport GCase to the lysosome.
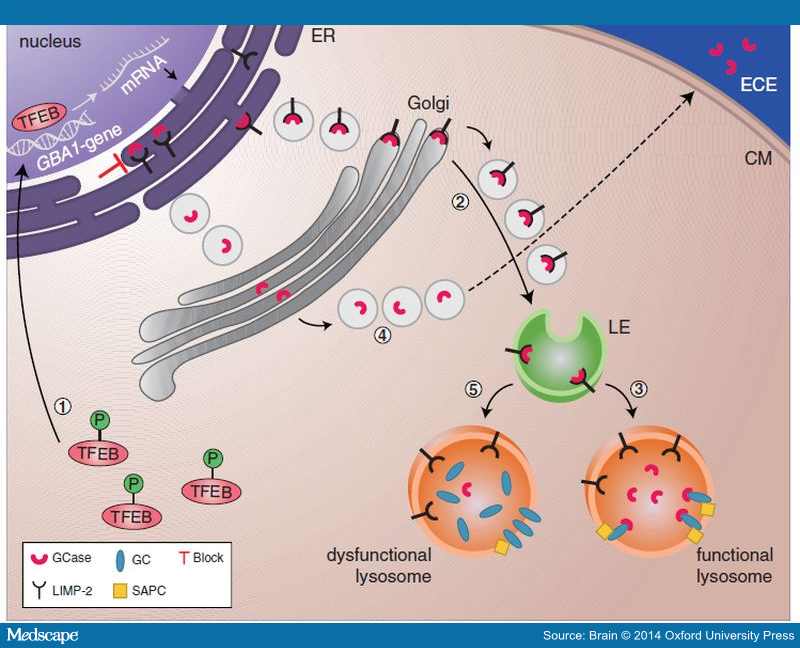 The function of LIMP2 in transporting GCase. Source: Medscape
The function of LIMP2 in transporting GCase. Source: Medscape
- Prof David Eidelberg (Feinstein Institute for Medical Research) will follow people with Parkinson’s (with and without GBA mutations) over a period of 18 months to assess progression as measured via clinical examinations and MRI scans. The goal of this project will be to develop an imaging biomarker.
- Dr Michele Matarazzo (University of British Columbia in Canada) will be looking for predictive biomarkers by assessing GBA genetic mutation carriers (with and without Parkinson’s) via clinical evaluations, blood tests, and MRI/PET scans (could be good for these last two to compare notes with the Rapsodi team in the UK).
- Prof David Vocadlo (Simon Fraser University in Canada) will be rapidly screening thousands of compounds to identify those that can increase GCase activity. He will then be testing the lead compounds in models of Parkinson’s.
- Dr Marlene Jacobson (Temple University) will be testing compounds that improve lysosomal function and increase GCase levels in skin cells from people with GBA-associated Parkinson’s.
- Dr Marta Martinez-Vicente (Vall d’Hebron Research Institute in Spain) will be conducting pre-clinical tests of compounds from Gain Therapeutics, which aim to stabilise GCase and help recover its activity.

Gain Therapeutics uses their site-directed enzyme enhancement therapy platform to identify compounds which are able to stabilize misfolded proteins AND restore the protein’s enzymatic activity. This video explains how this idea works:
- Dr Magdalene Moran at Rheostat Therapeutics will be looking at activators of TRPML1. Early experiments suggest that activation of this ion channel may help to fix lysosomal malfunctions, which are associated with GBA mutations (Click here to read more about TRPML1 & Parkinson’s).

That is a LOT of GBA/GCase-associated research being supported by the Silverstein Foundation and the Michael J Fox Foundation. The two organisations should be applauded for their collaborative efforts. And it will be very interesting to see the products of this research – hopefully not only novel therapeutics for GBA-associated Parkinson’s, but also a far more advanced understanding of the biology underlying this form of the condition.
So what does it all mean?
The battle against Parkinson’s is pulling together tremendous resources, across different funding groups, to help to focus leading edge technology, on an international stage. When reading through the list of projects being supported by the SIlverstein foundation and Michael J Fox Foundation, one can not help being impressed with the breadth of the areas being covered for just this one subtype of Parkinson’s. The projects cover everything from the genetics underlying the condtion, all the way through to new potential therapeutics.
I like this GBA-focused approach. Not only because it is acknowledging and targeting a sub-type of Parkinson’s (which is certainly the way ahead for now), but also because the fruits of this labour could have important implications for other subtypes of Parkinson’s (and possibly even Gaucher disease). By this I mean, a treatment that is fast-tracked for GBA-associated Parkinson’s might be found to also have benefits for other subtypes of Parkinson’s. And even if it doesn’t have an effect on other subtypes, that information may still teach us something about the differences in underlying biology between the different forms of Parkinson’s. Rather than testing a treatment on a randomly selected bunch of people with Parkinson’s, we are getting more sophisticated in our approach to new therapies.
And I am looking forward to learning more about GBA-associated Parkinson’s in the coming month as the Silverstein Foundation observes it’s second anniversary in March. Last year there was a string of annoucements associated with their first ‘birthday’ (Click here to read more about that). It will be interesting to see what news they have this year.
No expectations though – remember! No expectations.
The banner for today’s post was sourced from the Silverstein Foundation for Parkinson’s with GBA and the Michael J. Fox Foundation for Parkinson’s Research

Your website is very interesting and informative. There are however some inaccuracies in your summary of one of the articles about GBA.
It says “In this multi-research centre study, the investigators studied 1893 people with Parkinson’s. Of these 48 (2.5%) had one copy of a known GBA mutation, 117 (6.2%) had a non-GBA genetic variant that had previously been associated with Parkinson’s (such as mutations in genes like LRRK2 or Alpha Synuclein), and 28 (1.5%) carried genetic variants of unknown significance in the GBA gene. As I mentioned above, one of the most common GBA mutations associated with Parkinson’s is called ‘L444P‘ and this was also the most common GBA mutation observed in this study.”
This would be more accurate as follows:
In this multi-research centre study, the investigators studied 1893 people with Parkinson’s. Of these 171 (9.0%) had one copy of a known GBA mutation, of whom 121 (6.2%) had a genetic variant known to be associated with Parkinson’s but not causing Gaucher’s disease, and 50 (2.7%) had a genetic variant linked to both Parkinson’s and Gaucher’s. Another 28 (1.5%) carried genetic variants of unknown significance in the GBA gene. The most common GBA mutations associated with Parkinson’s in this study were ‘E362K‘, ‘T369M’ , and ‘L444P’.
LikeLike
Dear Dr Grosset,
Many thanks for your comment and suggested correction to the text – greatly appreciated. The text has now been changed as suggested.
Kind regards,
Simon
LikeLike