|
A recent study published by French, British and Swiss researchers has grabbed the attention of some readers. The report suggests that the inert/noble gas, Xenon, has powerful anti-dyskinetic properties in both mouse and primate models of Parkinson’s with L-DOPA-induced dyskinesias. Dyskinesias are involuntary movements that can develop over time with prolonged used of L-DOPA treatments. In today’s post, we will discuss what Xenon is, how it may be reducing dyskinesias, and we will consider some of the issues associated with using Xenon. |

Dyskinesia. Source: JAMA Neurology
There is a normal course of events following a diagnosis of Parkinson’s.
Yes, I am grossly over-generalising, and no, I’m not talking from personal experience, but just go with me on this for the sake of discussion.
First comes the shock of the actual diagnosis. For many it is devastating news – an event that changes the course of their future. For others, however, the words ‘you have Parkinson’s‘ can provide a strange sense of relief that their current situation has a name and gives them something to focus on.
This initial phase is usually followed by the roller coaster of various emotions (including disbelief, sadness, anger, denial). It depends on each individual.

The emotional rollercoaster. Source: Asklatisha
And then comes the period during which many will try to familiarise themselves with their new situation. They will read books, search online for information, join Facebook groups (Click here for a good one), etc.
That search for information often leads to awareness of some of the realities of the condition.
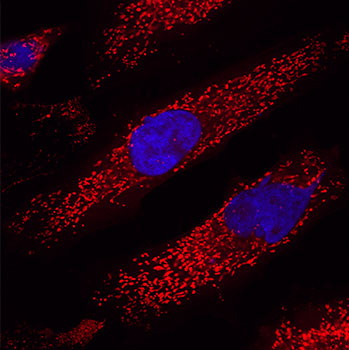
And one potential reality that causes concern for many people (especially for people with early onset Parkinson’s) is dyskinesias.
What are dyskinesias?