
|
Mitochondrial division inhibitor-1 (mdivi-1) is a small molecule drug that is demonstrating very impressive effects in preclinical models of Parkinson’s disease. With further research it could represent a potential future therapy for people with Parkinson’s disease, particularly those with genetic mutations affecting the mitochondria in their cells. What are mitochondria? In this post, we will explain what mitochondria are, how they may be involved in Parkinson’s disease, and we will discuss what the results of new research mean for future therapeutic strategies. |

Mitochondria are fascinating.
Utterly. Utterly. Fascinating.
On the most basic level, Mitochondria (mitochondrion, singular; from the Greek words mitos (thread) and chondros (granule)) are just tiny little bean-shaped structures within the cells in our body, and their primary function is to act as the power stations. They supply the bulk of energy that cells require to keep the lights on. This chemical form of energy produced by the mitochondria is called adenosine triphosphate (or ATP). Lots of mitochondria are required in each cell to help keep the cell alive (as is shown in the image below, which is showing just the mitochondria (red) and the nucleus (blue) of several cells).
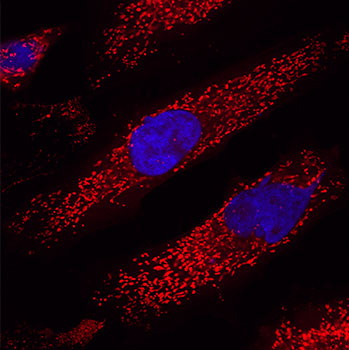
Lots of mitochondria (red) inside cells (nucleus in blue). Source: Clonetech
That’s the basic stuff – the general definition you will find in most text books on biology.
But let me ask you this:
How on earth did mitochondria come to be inside each cell and playing such a fundamental role?
I don’t know. Are you going to tell me?
No.
Why not?
Because we simply don’t know.
But understand this: Mitochondria are intruders.
They are not supposed to be there. There is no DNA in the nucleus of each cell that explains how to make a mitochondrion. They are foreign bodies inside the cell. Autonomous units that have their own agenda.
With regards to their origins, most theories suggest that mitochondria are actually an ancient bacteria of some sort.
How could we possibly know that?
We don’t. We are simply assuming it. But it’s a pretty good guess given all of the similarities between bacteria and mitochondria.
For example: mitochondria – like bacteria – contain their own DNA – that is to say, they have their own genetic instruction manual which is completely separate from the DNA that is sitting in the nucleus of almost every cell in your body. Mitochondrial DNA is inherited from your mother, and contains just 16,569 base pairs (think T,A,G & Cs). That DNA holds the instructions for just 37 genes.
In addition, the DNA of mitochondria – like that of bacteria – does not have histones. Histones are basic proteins which have positive charges that allow them to associate with DNA (which is negatively charged). They function as spools that the string-like DNA can wrap around. And their absence may explain why – again, like bacteria – histone-less mitochondrial DNA is maintained in a circular shape.
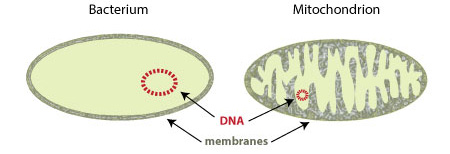
Source: Visionlearning
There are different theories of course about how mitochondria came to be there inside cells – the two main theories being the endosymbiosis theory and the autogenous theory – but the truth lies waaaaaaaay back in our distant evolutionary path. About four billion years ago to be un-precise. Back in the primordial soup that gave rise to all of life, mitochondria somehow snuck into an ancient early cell.
The basic idea is that at some point in that primordial soup phase, a bacterium of some sort was engulfed by a cell and having somehow survived that process, it decided that conditions were right for it to hang around. And subsequently, the two organisms decided that it was a favourable situation for both: the cell providing a rich environment of material for the mitochondria to thrive and divide, and the mitochondria providing energy in the form of ATP in exchange.
The situation between the two entities seems to work ok most of the time – there are hundreds (in some cases thousands) of mitochondria is each cell of your body, dividing every now and then as the need goes. And when a particular cell divides, a proportion of the mitochondria in that cell will go with each of the subsequent cells. To quote Sir Paul McCartney and Stevie Wonder, the cell and the mitochondria are like ‘Ebony and ivory, living together in perfect harmony‘.

Stevie Wonder andPaul McCartney. Source: Youtube
Except of course until something goes wrong…
And as with all good relationships, sometimes things go wrong.
Unfortunately, mitochondrial dysfunction is the root cause of many specific diseases, such as mitochondrial myopathy, ‘Neuropathy, ataxia, and retinitis pigmentosa‘ (also known as NARP syndrome), and Leigh syndrome.
And before we go on, I would just like to point out that mitochondria are not the only dysfunctional member of this relationship. There are conditions within which general cell dysfunction can affect mitochondria (such as Friedreich’s ataxia and many forms of cancer – click here and here to read more about this). I would hate to give mitochondria a bad name.
In the situation of mitochondrial dysfunction, however, much of it results from mutations in the mitochondrial DNA.
Are mutations in mitochondrial DNA associated with Parkinson’s disease?
Great question.
People have actually looked into this and there has been one report suggesting that a particular mutation in mitochondrial DNA can result in Parkinson’s-like features (called Parkinsonisms).
This is the research report:

Title: A novel mitochondrial 12SrRNA point mutation in parkinsonism, deafness, and neuropathy.
Authors: Thyagarajan D, Bressman S, Bruno C, Przedborski S, Shanske S, Lynch T, Fahn S, DiMauro S.
Journal: Ann Neurol. 2000 Nov;48(5):730-6.
PMID: 11079536
In this study, the investigators were presented with a woman of Italian background who at 45 years of age noticed a “resting tremor in the left hand, followed by stiffness of the left leg, a soft voice, a mildly unsteady gait, a weight loss of 5 lb, and word-finding difficulty”. At the start of her fourth decade, she had suffered depression and anxiety, followed by a transient episode of blurred vision at 43 years of age. All five of her maternal aunts or uncles had adult-onset deafness beginning in their 30s, and one of the aunts had developed Parkinson’s disease.
DNA sequencing revealed a novel mutation in the mitochondrial 12SrRNA gene within the affected individuals. The mutation appears to be very rare, as it was absent in the mitochondrial DNA from 270 controls (of Japanese, caucasian, and African-American origins) and in 20 people with idiopathic Parkinson’s disease. The timing of the deafness and Parkinsonisms is curious though.
Does age have an effect on mitochondria?
There is an age-dependent increase in the number of the common mitochondria DNA mutations. As we get older and mitochondria divide, the ‘intruders’ seems to incur more mistakes in their DNA (Click here to read more on this).
With regards to Parkinson’s disease specifically, in 2006 two independent research groups published two reports side-by-side demonstrating that there is a high level of mitochondrial DNA disruptions in the dopamine neurons, which are so badly affected by Parkinson’s disease.
This was the first report:

Title: High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease.
Authors: Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM.
Journal: Nat Genet. 2006 May;38(5):515-7.
PMID: 16604074
In this first study, the researchers found very high levels of mutations in mitochondrial DNA in the dopamine neurons in the substantia nigra (a region of the brain vulnerable to Parkinson’s disease) of normal healthy aged control brains (approximately 43.3% of mitochondria were affected). But this level of affected mitochondria is increased in individuals with Parkinson disease (52.3%). The mutations are also somatic – that is to say that the mutations have been collected during the years of life, rather than the individuals being born with them.
And the second research report reported something similar:
Title: Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons.
Authors: Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K.
Journal:Nat Genet. 2006 May;38(5):518-20.
PMID: 16604072
In this study, the investigators also found a very high levels (approx. 60%) of mutations in mitochondrial DNA in the substantia nigra of aged brains, suggesting that there is a natural reduction in the quality of mitochondrial DNA over time in the dopamine neurons – which could be making them more vulnerable to disease or environmental toxins.
Ok. But can we treat dysfunctional mitochondria?
There is a lot of on-going research investigating how we can better control mitochondrial activity. In particular, researchers have conducted large screening studies that have searched for proteins or drugs that can stop mitochondria from dividing. Mitochondria can divide or fusion together depending on their needs or on what is happening around them.These processes can play important roles in the regulation of programmed cell death (also called apoptosis). Proteins that are involved in mitochondrial fusion help to reduce the chance of apoptosis, by inhibiting release of a protein called ‘cytochrome c’ from mitochondria. The release of cytochrome c is bad news for a cell. Proteins involved in mitochondrial division are known to promote apoptosis (although the mechanism of this is not clear).
This is why a lot of research has been conduced on finding the proteins or drugs that might help regulate mitochondrial division and fusion.
Back in 2008, one of these screens identified one particularly interesting drug:

Title: Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization.
Authors: Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J.
Journal: Dev Cell. 2008 Feb;14(2):193-204.
PMID: 18267088 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators screened approximately 23,000 chemical compounds for their ability to inhibit mitochondrial division in yeast. Yeast is a single-celled microorganism, that has become one of the most centrally important model organisms used in modern biological research. It is one of the most thoroughly researched organisms on the planet (we know more about the biology of yeast than we do about ourselves!).

Yeast cells. Source: NewEuropeans
The biology of these cells is very similar to our own and this is why they are being used in studies screening new drugs for different human conditions, including neurodegenerative diseases (Click here to read a previous post about this).
The researchers identified three drugs with potent mitochondrial division inhibitory activity, and they decided to further investigate the most efficacious. It was called mdivi-1 (for mitochondrial division inhibitor no.1). Interestingly, this drug not only worked in yeast cells but also mammalian cells, increasing its potential utility. But it was what the research found next that really caught their attention: mdivi-1 could block apoptosis by inhibiting the outer membrane of mitochondria from opening up and releasing toxic chemicals that encourage the cell is die.
mdivi-1 blocked mitochondrial division and cell apoptosis by inhibiting a protein called dynamin-related GTPase 1 (DRP1)
The investigators concluded that “mdivi-1 represents a novel class of therapeutics for stroke, myocardial infarction, and neurodegenerative diseases“.
Interesting, but what does all of this have to do with Parkinson’s disease?
This is Prof Kim Tieu:

Prof Kim Tieu. Source: FIU
He’s awesome. Formerly at Plymouth University (UK) now at Florida International University (USA), his lab has been working on the DRP1 inhibitor drug mdivi-1 within the field of Parkinson’s disease, and they have made some very interesting discoveries.
The first research report on this topic that they published was this one:

Title: Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1.
Authors: Cui M, Tang X, Christian WV, Yoon Y, Tieu K.
Journal: J Biol Chem. 2010 Apr 9;285(15):11740-52.
PMID: 20164189 (This article is OPEN ACCESS if you would like to read it)
In this study, Prof Tieu and his colleagues were interested in the activity of two Parkinson’s disease associated proteins, called PINK1 and PARKIN. About 10% of cases of Parkinson’s disease can be attributed to genetic mutations in particular genes. PINK1 and PARKIN are two of those genes. People with certain mutations in the PINK1 or PARKIN gene are more vulnerable to developing an early onset form of Parkinson’s disease.
In normal, healthy cells, the PINK1 protein is absorbed by mitochondria and eventually degraded. In unhealthy cells, however, this process is inhibited and PINK1 starts to accumulate on the outer surface of the mitochondria. There, it starts grabbing the PARKIN protein and this pairing is a signal to the cell that this particular mitochondria is not healthy and needs to be removed.

Pink1 and Parkin in normal (right) and unhealthy (left) situations. Source: Hindawi
The process by which mitochondria are removed is called autophagy. Autophagy is an absolutely essential function in a cell. Without it, old proteins will pile up making the cell sick and eventually it dies. Through the process of autophagy, the cell can break down the old protein, clearing the way for fresh new proteins to do their job.
Think of autophagy as the waste disposal/recycling process of the cell.
In the absence of normally functioning PINK1 and PARKIN – as is the case in some people with Parkinson’s disease who have genetic mutations in these genes – we believe that sick/damaged mitochondria start to pile up and are not disposed of appropriately. This results in the cell dying.
Prof Tieu and his team found PINK1 has pro-fusion and inhibition of division functions on mitochondria. Mutant forms of the PINK1 protein (L347P and W437X) increasing the number of mitochondrial division proteins, leading to an excessive of dysfunctional and fragmented mitochondria. And they saw the same effect when they simply reduced normal levels of PINK1 protein, while increasing levels of Pink1 resulted in an increase in mitochondrial fusion. These findings reenforced the idea that PINK1 has pro-fusion properties.
When the researchers reduced the levels of the other Parkinson’s associated protein, PARKIN, they observed similar effects – an increase in the proteins involved in mitochondrial division, resulting in dysfunctional and fragmented mitochondria – suggesting that PINK1 and PARKIN maintain proper mitochondrial function via the machinery of division and fusion.
Next they tested the drug mdivi-1 on the cells with mutant PINK1 protein. They found that mdivi-1 treatment inhibited both the structural and functional mitochondrial defects when compared to non-treated cells with mutant PINK1 protein. And they concluded that the drug mdivi-1 was “a potential novel therapeutic avenue for Parkinson disease”.
This first study was followed up several years later by another research report further investigating mdivi-1 in two mouse model of Parkinson’s disease:

Title: Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo.
Authors: Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, Zhuang X, Bowers WJ, Tieu K.
Journal: Nat Commun. 2014 Nov 5;5:5244.
PMID: 25370169 (This article is OPEN ACCESS if you would like to read it)
In this study, the researchers treated genetically engineered mice (which have had their PINK1 gene mutated) with the mdivi-1 drug. These PINK1 mutant mice have dysfunctional dopamine release, which mdivi-1 treatment helped to correct. This result is important because it also demonstrates that mdivi-1 can enter the brain (passing through the protective layer that is called the blood-brain-barrier). Next the investigators assessed the ability of mdivi-1 to protect dopamine neurons in the brain from a toxin (MPTP) that causes Parkinson’s-like features. It did! And it helped to restore normal dopamine processing in the brain.
Given this result, the researchers decided to investigate the effect of mdivi-1 on a more Parkinson’s disease relevant animal model – that is, one in which the Parkinson’s associated protein alpha synuclein is over produced.
The results of that study have just been published:

Title: Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease.
Authors: Bido S, Soria FN, Fan RZ, Bezard E, Tieu K.
Journal: Sci Rep. 2017 Aug 8;7(1):7495. doi: 10.1038/s41598-017-07181-0.
PMID: 28790323 (This article is OPEN ACCESS if you would like to read it)
In this study, Prof Tieu and his colleagues evaluated whether blocking of mitochondrial division with mdivi-1 was neuroprotective against high levels of mutant human alpha synuclein (A53T) protein in rodents. They found that mdivi-1 was again very effective at reducing neurodegeneration in the dopamine neurons, but it also reduced the clustering (or aggregation) of alpha synuclein and normalised motor function in the rats. They concluded that “strategies of blocking Drp1 function hold great promise to novel therapies”.
Wow, this is great! Is mdivi-1 heading for the clinic?
- There is no patent on it.
- There could be side effects.
On the former, without a patent providing exclusive rights to the drug, there is little chance that a biotech or large pharmaceutical company will spend the millions of dollars required for taking the drug through the clinical trial process. If the biotech company can not be promised the sole right to generate profits on expense of clinical trials, there isn’t much chance that a drug will be taken forward for clinical testing.
And this is, unfortunately, becoming a common theme. We now have numerous experimental drugs that have been placed in the public domain (rendering them un-patentable), which should be being tested in the clinic for Parkinson’s disease (recently we looked at potent anti-dyskinesia drugs UWA-101 and UWA-121 which are not patented and also not heading to clinic – Click here to read that post). What we require is a new business model that can prioritise these un-patented drugs (it’s just an idea D.A.J, but maybe something to think about). The alternative is to wait for derivatives of mdivi-1 that are patentable, but this will require a lot more time and research.
There is, however, a patent protecting a different mdivi-1-like drug, called ‘P110’. This patent is controlled by a small university in California called the Leland Stanford Junior (colloquially called ‘the Farm’, though you might know it as Stanford University). And this drug is also being tested in models of Parkinson’s disease:

Title: Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP.
Authors: Filichia E, Hoffer B, Qi X, Luo Y.
Journal: Sci Rep. 2016 Sep 13;6:32656.
PMID: 27619562 (This article is OPEN ACCESS if you would like to read it)
In this study, the investigators assessed the ability of the GRP1 inhibitor, P110 to protect dopamine neurons on a mouse model of Parkinson’s disease. The mice were treated with a toxin (MPTP) that targets dopamine neurons, and the researchers found that the mice treated with P110 had significantly reduced loss of dopamine neurons and improvements in their behavioural deficits. Curiously, the investigators reported that activation of the helper cells in the brain (the microglia) was not affected by P110 treatment. Rather the neuroprotective effect was due to reduced levels of apoptosis associated proteins.
These researchers have also demonstrated positive effects with P110 treatment in cells that have mutations in a Parkinson’s associated gene called Leucine-rich repeat kinase 2 (LRRK2) (Click here to read that research report).
But while all of this might sound really good (a patented drug similar to mdivi-1 that has beneficial effects in models of Parkinson’s disease), there is still the issue of side effects.
These drugs – mdivi-1 and P110 – are fiddling with one of the most important components of a cell: the power station. If we start treating people with a drug like this on a long-term basis, there could be the potential for serious side effects (not only in the brain, but elsewhere in the body as well). In the discussion of their latest research report, Prof Tieu and colleagues address this, noting that “blocking mitochondrial fission as a therapeutic strategy” may lead to “side effects, because a balance of [mitochondrial] fission and fusion is necessary for the maintenance of neuronal function.” While the previous research suggests that normal mice display no abnormalities after ten weeks of mdivi-1 treatment, what happens during longer term treatment regimes? Such questions will need to be assessed in further research.
What does it all mean?
A great deal of research is investigating the role of different drugs/proteins in mitochondrial division and fusion. Some of these agents appear to influence the balance of life and death in cells by inhibiting the ability of mitochondria to divide. Two of these drugs have demonstrated very positive effects in models of Parkinson’s disease. Whether such drugs can be taken into the clinic is yet to be determined, particularly given the possibility of off-target side effects. But since research indicates that mitochondria are vulnerable to the effects of age and Parkinson’s disease, therapies designed around this approach are worthy of further research.
The banner for today’s post was sourced from NPR

The wow-factor is huge here, Simon…great teaching that I’ll be chewing on for some time!
LikeLike
Hi Lisa,
Thanks for the comment. Glad you liked the post. It is yet another approach that research is taking to beat this Parkinson’s beast. I worry that there may be a lot of off target effects with this sort of drug, but perhaps with more research we will be able to resolve that.
Enjoy your weekend,
Simon
LikeLike
Hi Simon
Thanks for this article. All this research over the last several years indicating decreasing DRP1 protein to be beneficial for stopping Parkinson’s and maintenance of mitochondria. But this most recent study below indicating in other organisms and cells just the opposite with huge benefits in life extension by increasing DRP1. What to make of this?
Mike
https://www.nature.com/articles/s41467-017-00525-4
LikeLike
Hi Mike,
Thanks for the comment – glad you liked the post. I mentioned this report in the twitter feed the other day and I wondered if anyone would notice. I am scratching my head trying to understand that study. It is set in the context of healthy, normal flies, but why would a transient induction of Drp1 – in midlife – prolong lifespan? Usually mitochondrial fission is associated with the increased chance of nasty things happening. I would be curious to see how this plays out in normal mice. Very curious.
Kind regards,
Simon
LikeLike