
|
A new research report has been published this week which may point not only towards a new understanding of the biology of Parkinson’s, but also to potentially novel therapies which are clinically available. These exciting new findings involve a DNA repair mechanism called ‘poly ADP ribose polymerase’ (or simply PARP) and a process of cell death called Parthanatos. Biotech companies have developed PARP inhibitors which have been reported to rescue models of Parkinson’s. With a bit of tweaking, this class of drugs could potentially be re-purposed for Parkinson’s. In today’s post, we will look at what PARP is, explain how PARP inhibitors work, review what previous PD research has been conducted on this topic, evaluate the new report, and consider what it means for the Parkinson’s community (Spoiler alert: this will be a long post!).
|

Source: Quotefancy
Ah, the good old days!
Remember them. Way back before Netflix. When life was sooo much easier.
You know what I’m talking about.
Back when biology was simple. Remember when DNA gave rise to RNA and RNA gave rise to protein, and that was it. Simpler times they were. Now, everything is so much more complicated. We have all manner of ‘regulatory RNA’, epigentics, splice variants, and let’s not get started on the labyrinthian world of protein folding.
Oh, how I long for the good old days.
Back when a cell could only die one of two ways: apoptosis (a carefully controlled programmed manner of death) and necrosis (cell death by injury):
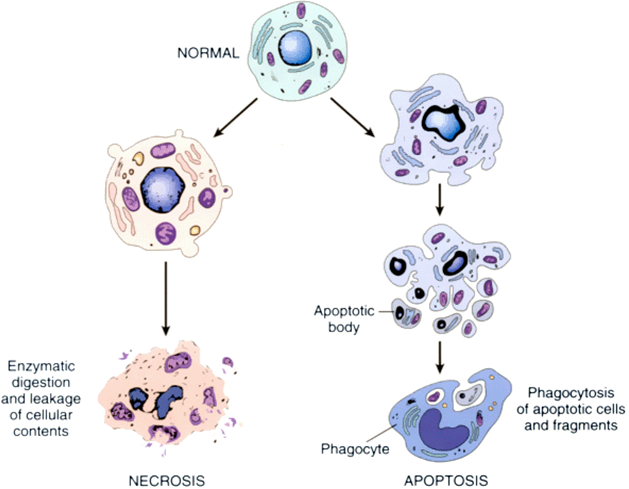
Source: Researchgate
Now life is too complicated and complex beyond reason or imagination.
Let’s just take the example of cell death that I mentioned above: over the past decade, the Nomenclature Committee on Cell Death (or NCCD – I kid you not there is actually a committee for this) has written up guidelines for the definition/interpretation of ‘cell death’. And as part of that effort they have decided that there are now at least 12 (yes, 12) different ways a cell can die:
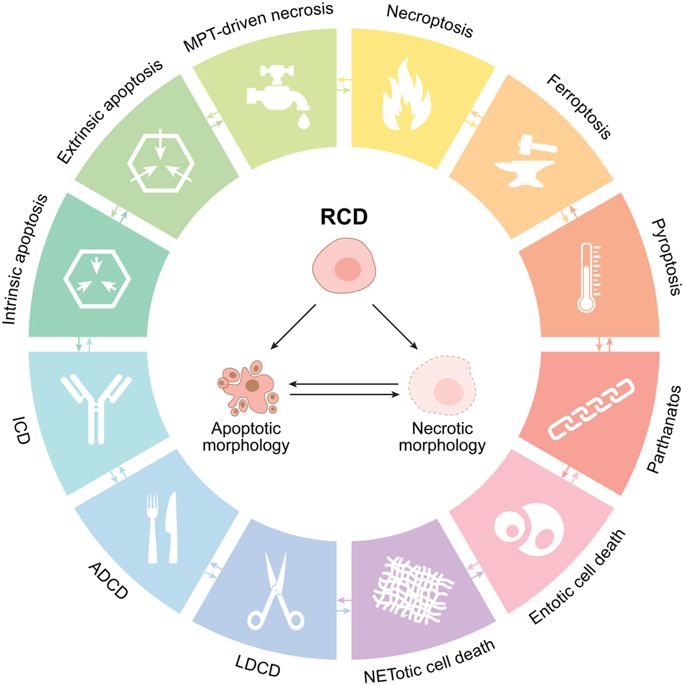 Source: Nature
Source: Nature
For those of who are interested in reading more about all of these different kinds of cell death, click here to read NCCD committee’s most recent recommendations which were updated this year (2018). Some riveting betime reading.
Which form of cell death applies to Parkinson’s?
Now that’s a really good question!
One that has been studied and the source of debate for a very long time.
To be fair, we don’t really know. But fascinating new research published this week suggests that the Parthanatos pathway could be involved in the cell death associated with Parkinson’s.
What is Parthanatos?
First proposed in 2009 (Source), Parthanatos (which is derived from the Greek Θάνατος or Thanatos – literally the personification of death in Greek mythology) is a form of cell death that involves very specific biological pathways in a cell.
 Thanatos. Source: Wikipedia
Thanatos. Source: Wikipedia
The Parthanatos form of cell death is characterised by two main features:
- the excessive production of poly-ADP ribose polymer
- the release of a protein called ‘apoptosis-inducing factor’ from the mitochondria
What is poly-ADP ribose polymer?
Poly-ADP ribose (or simply PAR) is part of a DNA repair mechanism in cells.
When a cell is put under stress, there will quite often be damage to the DNA in that cell.

Different forms of stress that lead to DNA damage. Source: Sierraoncology
And given the importance of DNA, mother nature has very wisely devised several very clever systems of DNA repair to maintain the integrity of this precious molecule.
One of those DNA repair mechanisms involves a protein called PARP.
And what is PARP?
Poly (ADP-ribose) polymerase (or PARP) is a family of proteins involved in a number of cellular processes such as DNA repair, genomic stability, and programmed cell death. There are currently 17 different types of PARP.
The research discussed in today’s post will be largely focused on PARP1, but for simplicity sake I will only refer to it as PARP.
The main function of PARP in cells is to detect DNA damage and initiate a response.
When damaged DNA is detected, PARP will bind to the DNA and begin to synthesise chains of poly-ADP ribose (or PAR which we mentioned above). These tendrils of PAR serve as a signalling mechanism for DNA-repairing enzymes, making them aware of the damage and recruiting their help to fix it.
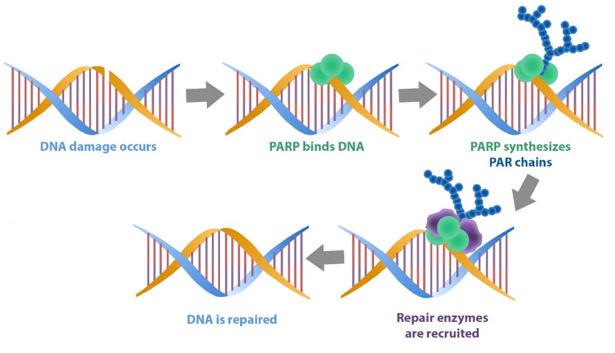 PARP initiating DNA repair mechanisms. Source: bpsbioscience
PARP initiating DNA repair mechanisms. Source: bpsbioscience
This is a very good system if the DNA damage is not too bad and if PARP activity is kept under strict control. But problems start to arise when PARP gets a bit carried away and becomes over activated, causing an accumulation of PAR.
You see, if there is too much PAR floating around with nothing to do, it will start to cause trouble.
What kind of trouble?
One of the ways PAR can wreak havoc is by spilling out from nucleus of the cell (where it is usually found) and into the surrounding internal world of the cell. Once outside of the cell nucleus, PAR will head over to places it shouldn’t be, such as the mitochondria.
What are mitochondria?
Mitochondria are the power stations of each cell. They help to keep the lights on. Without them, the party is over and the cell dies.

Mitochondria and their location in the cell. Source: NCBI
Mitochondria can also have a powerful influence of cell survival in other ways.
Theses small bean shaped objects not only provide energy for the cell, but they can also release signalling proteins which can instruct the cell what to do.
For example, when PAR reaches the mitochondria it will bind to a protein called apoptosis-inducing factor, which is a protein that (as the ‘label-on-the-can’ suggests) induces apoptosis, a type of programmed cell death.
 Source: Sciencedirect
Source: Sciencedirect
PAR entering the mitochondria causes apoptosis-inducing factor to be released from mitochondria, and once unleashed apoptosis-inducing factor makes its way to the nucleus where it will start to activate regions of DNA that are involved with shutting down the cell (Click here to read more about this).
Left unchecked, this process will rapidly lead to the cell dying.
And this is Parthanatos in a nut shell (increase in PAR levels, causing apoptosis-inducing factor to be released from mitochondria, resulting in cell death).
What does PARP or Parthanatos have to do with Parkinson’s though?
In 1998, this research report was published:
 Title: Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation.
Title: Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation.
Authors: Lo EH, Bosque-Hamilton P, Meng W.
Journal: Stroke. 1998 Apr;29(4):830-6.
PMID: 9550519 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers noticed that PARP levels increase dramatically in the brain after an ischemic injury (stroke), and they wondered whether blocking PARP would help or hinder the recovery. They modelled stroke in two groups of rats (one group was treated with the PARP inhibitor 3-AB and the other group were used as controls), and they analysed their brains 24 hours after the induction of stroke. The treated group were given the PARP inhibitor immediately after the stroke was induced.
The investigators found that treating the animals with the PARP inhibitor significantly reduced the area of damage in the brain (the reduction was over 50%).
And this finding got other researchers asking whether PARP could be involved in other neurological conditions, including Parkinson’s.
Which led to this report being published in 1999:
 Title: Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism.
Title: Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism.
Authors: Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM.
Journal: Proc Natl Acad Sci U S A. 1999 May 11;96(10):5774-5779.
PMID: 10318960 (This report is OPEN ACCESS if you would like to read it)
The researchers found that shorty after treating mice with a neurotoxin (MPTP) which kills dopamine neurons, the dopamine neurons in the brain would produce high levels of PARP (in the image below, the dopamine neurons on the substantia nigra pars compacta (SNpc) are visible because the protein PARP has been stained red):
 Source: PMC
Source: PMC
The researchers next genetically engineered some mice that do not produce PARP and they conducted an experiment in which the no-PARP mice and normal mice were treated with the MPTP neurotoxin. The investigators found that while the normal mice lost more than half of their dopamine neurons, the neurotoxin had little (if any) effect on the dopamine neurons in the no-PARP mice.
Next, these researchers wanted to better understand the process by which this PARP-associated cell death was occuring, and that research lead to this report:
 Title: A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism.
Title: A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism.
Authors: Mandir AS, Simbulan-Rosenthal CM, Poitras MF, Lumpkin JR, Dawson VL, Smulson ME, Dawson TM.
Journal: J Neurochem. 2002 Oct;83(1):186-92.
PMID: 12358742 (This report is OPEN ACCESS if you would like to read it)
In this study, the investigators found that following the neurotoxin treatment, PARP was interacting with another protein called p53.
What is p53?
p53 (or TP53) is a protein that has three major functions:
- controlling cell division
- DNA repair
- Apoptosis (or cell death).
p53 performs these functions as a transcriptional activator (that is a protein that binds to DNA and helps produce RNA (the process of transcription) – see our previous post explaining this).

p53 protein structure, bound to DNA (in gold). Source: Wikipedia
In regulating the cell division, p53 prevents cells from dividing too much and in this role it is known as a tumour suppression – it suppresses the emergence of cancerous tumours. Genetic mutations in the p53 gene can result in run away cell division, and (surprise!) as many as 50% of all human tumours contain mutations in the p53 gene.

Cancer vs no cancer. Source: Khan Academy
With regards to DNA repair, p53 is sometimes called “the guardian of the genome” as it prevents mutations and helps to conserve stability in the genome. This function also serves to prevent the development of cancer, by helping to repair potentially cancer causing mutations. In this role, p53 is known as a tumour suppressor – a mutation in the p53 gene results in a loss of this tumor suppression.
And finally, in cell death, p53 plays a critical role in telling a cell when to die. And (continuing with the cancer theme), if there is a mutation in the p53 gene, fewer cells will be told to die – increasing the risk of cancer occurring. And in this role p53 is known as a tumour suppression. But in the absence of a mutation in the p53 gene or PARKIN to reduce the activity of the gene, p53 levels will gradually rise and instruct the cell to initiate apoptosis.
What does PARP do when it interacts with p53?
When PARP interacts with p53, it stablises the p53 protein – which is no good.
p53 usually has a very short life span, and by stablising p53, PARP is probably seeking its help in DNA repair. But this stablisation is also increasing the risk that p53 will encourage cells to die. And the researchers suggested that it could be this influence of PARP on p53 that may underlie the mechanisms of neurotoxin-induced cell death.
This finding was made before Parthanatos had been discovered, and it highlights another potential mechanism by which over active PARP could be detrimental to cells.
And the finding that PARP over-activation is involved in cell death has been replicated by other independent research groups. In addition, some of those groups have extended the research by looking at drugs that can inhibit PARP in models of Parkinson’s.
Such as this study:
 Title: Neuroprotective effects of a novel poly(ADP-ribose) polymerase-1 inhibitor, 2-[3-[4-(4-chlorophenyl)-1-piperazinyl] propyl]-4(3H)-quinazolinone (FR255595), in an in vitro model of cell death and in mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease.
Title: Neuroprotective effects of a novel poly(ADP-ribose) polymerase-1 inhibitor, 2-[3-[4-(4-chlorophenyl)-1-piperazinyl] propyl]-4(3H)-quinazolinone (FR255595), in an in vitro model of cell death and in mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease.
Authors: Iwashita A, Yamazaki S, Mihara K, Hattori K, Yamamoto H, Ishida J, Matsuoka N, Mutoh S.
Journal: J Pharmacol Exp Ther. 2004 Jun;309(3):1067-78.
PMID: 14985416 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers noted that over activation of PARP can lead to severe, irreversible depletion of the levels of NAD and ATP.
What are NAD and ATP pools?
Nicotinamide adenine dinucleotide (or NAD) is a protein that plays a very critical role in a wide range of cellular reactions. Importantly, it is essential for the continued production of energy by the mitochondria in cells.
Mitochondria convert nutrients from food into Adenosine Triphosphate (or ATP). ATP is the fuel which cells run on, and NAD is required for the production of ATP.
We have discussed NAD in a previous post on the SoPD website (Click here to read that post), and it is critical for the normal functioning of cells. Severely reducing levels of NAD can be very bad news for cells.
But here is the thing: NAD is also required for PARP to do its DNA repair job.
You see when PARP binds to DNA it uses NAD to produce the chains of PAR that we were talking about above.
 The role of NAD in PARP DNA repair. Source: Bpsbioscience
The role of NAD in PARP DNA repair. Source: Bpsbioscience
So if PARP becomes over activated in a cell, it can quickly reduce the supply of NAD, causing further trouble for cells.
The researchers in this particular report wanted to determine if blocking PARP activity could help to rescue a neurotoxin (MPTP) model of Parkinson’s, and restore NAD/ATP levels. They used a PARP inhibitor called FR261529, which was originally developed by Fujisawa Pharmaceutical Co., Ltd (which is now part of Astellas Pharma), but this drug is no longer being developed.
They injected mice with the PARP inhibitor 1 hour before giving them the neurotoxin and they found that it exerted a powerful neuroprotective effect and restored levels of NAD, which lead the researchers to conclude that PARP inhibition “could be an attractive candidate for several neurodegenerative disorders, including PD“.
This same research group expanded on these results – publishing a second research report just 4 months later (Click here to read that research report).
And other research groups have also replicated these results (Click here, here, and here to read some examples).
Interesting. But these are all models of Parkinson’s. Is PARP actully relevant in people with Parkinson’s?
Well, about the same time as these PARP inhibitor reports were being published, researcher also reported increased levels of PARP in the dopamine neurons in the postmortem brain of people with Parkinson’s (Click here to read more about this). In addition, there have been reports suggesting that genetic mutations in the PARP genes may actually protect people from developing Parkinson’s (Click here and here to read more about this).
Some research groups have also found that inhibition of PARP reduces alpha synuclein levels (Click here to read more about this).
And then this week, this research report was published:
 Title: Poly (ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease
Title: Poly (ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease
Authors: Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, Andrabi SA, Qi C, Poirier GG, Pletnikova O, Troncoso JC, Bekris LM, Leverenz JB, Pantelyat A, Ko HS, Rosenthal LS, Dawson TM, Dawson VL.
Journal: Science, 2018 Nov 2;362(6414). pii: eaat8407.
PMID: 30385548
In this study the researchers treated neurons grown in cell culture to preformed alpha synuclein fibrils.
What is alpha synclein fibrils?
Alpha synuclein is one of the most abundant proteins in our brains – making up about 1% of all the proteins floating around in each neuron in your head – and it is a very well studied protein (with over 9700 research reports listed on the Pubmed search engine with the key words ‘alpha synuclein’).
When alpha synuclein protein is produced by a cell, it normally referred as an ‘unfolded protein’, in that is does not really have a defined structure. When it is first produced, alpha synuclein will look something like this:
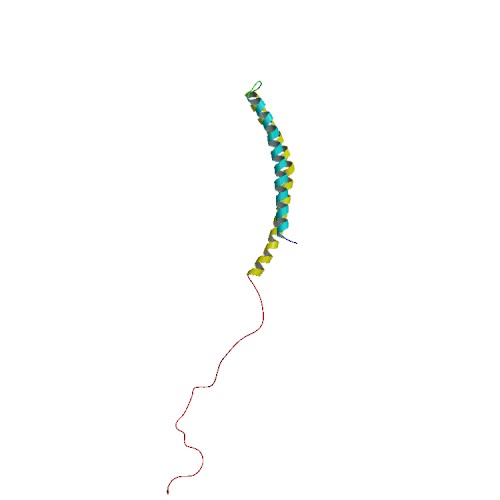
Alpha synuclein. Source: Wikipedia
In this form, alpha synuclein is considered a monomer – which is a single molecule, just one copy of the protein. It is capable of binding to other molecules, and when it binds to other alpha synuclein proteins, they form what is called an oligomer (a collection of monomers). And these oligomers can have different structures.
In Parkinson’s, alpha synuclein also binds (or aggregates) to form what are called ‘fibrils’.

Microscopic images of monomers, oligomers and fibrils. Source: Brain
And it is believed that these oligomer and fibril forms of alpha synuclein protein may go on to produce the Lewy bodies that characterise the Parkinsonian brain.
 Parkinson’s associated alpha synuclein. Source: Nature
Parkinson’s associated alpha synuclein. Source: Nature
By treating neurons grown in cell culture with preformed alpha synuclein fibrils, the researchers noted an increase in levels of both PARP and PAR, as well as an increase in cell death. When they treated these cells with PARP inhibitors, they found that the levels of PARP, PAR, and cell death all dropped dramatically.
Genetically removing PARP from the cells (using CRISPR gene editing technology) also protected neurons from the toxic effects of preformed alpha synuclein fibrils. These (and other experiments) suggested that preformed alpha synuclein fibrils were primarily killing neurons via parthanatos.
To confirm this result, the investigators next injected preformed alpha synuclein fibrils into both normal mice and mice that were genetically engineered to have no PARP.
And guess what?

Six months after being injected with preformed alpha synuclein fibrils, the dopamine neuron population in the normal mice was reduced by approximately 50%, but the mice with no PARP exhibited no dopamine cell loss. The researchers repeated this experiment with two groups of normal mice, but they treated one group with PARP inhibitor drugs. Again, after 6 months the PARP inhibitor treated mice exhibited little if any cell loss compared to their untreated counterparts (who suffered 50% dopamine neuron loss).
Now, because the rise in levels of PARP were associated with an increase in levels of PAR (when cells were treated with preformed alpha synuclein fibrils), the researchers asked what effect PAR itself may be having.
When they exposed normal human alpha synuclein protein to PAR in a solution, the investigators reported a marked acceleration of protein aggregation. In addition, when they looked at the brains of mice injected with preformed alpha synuclein fibrils, they found that 20% of alpha synuclein was bound to PAR.
On top of this result, exposing both normal cells and cells with no PARP to increased levels of PAR resulted in increased aggregation of alpha synuclein – which suggested to the researchers that it is PAR and not PARP that directly increases levels of alpha synuclein aggregation. By treating the cells with PARP inhibitors, the levels of PAR were also significantly reduced, which in turn resulted in less alpha synuclein aggregation.
This increase in PAR-induced alpha synuclein aggregation resulted in an raised levels of cell death in neurons grown in culture. And in the absence of alpha synuclein, they found that PAR was not toxic (even at very high levels).
Further investigations suggested that exposing preformed alpha synuclein fibrils to PAR was causing them to adopt an even more toxic state. And this demonstrated itself further when the researchers injected either PAR exposed preformed alpha synuclein fibrils or just normal preformed alpha synuclein fibrils into mice: the mice injected with PAR exposed preformed alpha synuclein fibrils had a more rapid loss of dopamine neurons and behavioural/motor problems.
Whoa! But has anyone ever looked in humans? Are there increased levels of PAR in humans?
Yes, this study was published a few years back:
 Title: Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss.
Title: Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss.
Authors: Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D, Jiang H, Kang SU, Lee BD, Kang HC, Kim D, Tessarollo L, Dawson VL, Dawson TM.
Journal: Nat Neurosci. 2013 Oct;16(10):1392-400.
PMID: 23974709 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers found that PAR levels were increased in the midbrain of people with Parkinson’s (compared to healthy control samples). This analysis was conducted on post mortem sections of midbrain – and the midbrain is where the bulk of the dopamine neurons reside in the human brain.
In the report from this week which we are reviewing today, the researchers also looked at PAR levels in people with Parkinson’s. For their analysis, the investigators used samples collected from two independent groups of people with Parkinson’s (collectively over 100 individuals with PD and 61 controls), and they found increased levels of PAR in the cerebrospinal fluid (this is the fluid that the brain sits in). Intriguingly, one of those cohorts actually demonstrates an association between PAR levels in the cerebrospinal fluid and disease duration – meaning, the longer each person has had Parkinson’s, the higher their levels of PAR.
The researchers summarised their report with the following conclusions:
- Alpha synuclein is killing cells by activating PARP (via the parthanatos cell death pathway)
- PARP activation leads to increased levels of PAR which accelerate alpha synuclein aggregation (a feed-forward cycle)
- PAR levels are increased in the brains of people with Parkinson’s
Given all of these results, the investigators suggested that clinically available PARP inhibitors should be considered for clinical testing in Parkinson’s
Wow! Has a PARP inhibitor ever been tested in Parkinson’s?
Not that I am aware of,… but we can assume that this is going to change very shortly.
PARP inhibitors are currently used in the treatment of certain cancers.
 PARP inhibition in cancer. Source: Parp-inhibitors
PARP inhibition in cancer. Source: Parp-inhibitors
The most common treatment for many cancers is chemotherapy, which functions by causing fatal DNA damage in cancer tumor cells. Key DNA repair pathways (like PARP), however, are hyperactive in a lot of cancers which results in increased resistance to chemotherapy treatment. But by inhibiting the DNA repair mechanism – by blocking PARP – researchers can potentiate the effect of chemotherapy, and increase the chances of killing off the tumor cells.
And this has resulted in the development of numerous PARP inhibitors by multiple pharmaceutical companies for clinical use in cancer.
Repurposing this class of drugs for Parkinson’s will be difficult, however, as most of the available PARP inhibitors have very poor penetration of the brain. They all have a lot of trouble getting across the blood-brain-barrier – the protective membrane surrounding our brains. And simply using higher doses of these drugs to increase levels actually entering the brain is a non-starter, due to the side effects associated with the PARP inhibitors.
Side effects include nausea (generally in 50% of cases), fatigue (33%), anemia (low hemoglobin levels in the blood), vomiting, and neutropenia (low level of neutrophils, white blood cells important to fighting off infections).
Thus, we will need to wait for compounds that do access the brain before such clinical trials can begin. Ideally, if those drugs could easily access the brain, perhaps we would be able to lower the dose and reduce the chances of negative side effects.
But the good news is that this was an area of VERY active interest for the research field even before this new report was published – Click here to read a rather comprehensive review (written in early 2017) of repurposing PARP inhibitors for the therapy of non‐oncological conditions.
So what does it all mean?
This is Profs Ted and Valina Dawson:
 Source: Hopkinsmedicine
Source: Hopkinsmedicine
They are the husband and wife team at John Hopkins University behind not only much of the research presented in today’s post, but also providers of a lot of the research supporting other experimental therapeutic approaches for Parkinson’s (including Nilotinib and Exenatide). In addition, they were the first to define the Parthanatos cell death pathway (Source).
Two champions of the cause who deserve a special mention here.
Their labs have recently published a research report (which we have reviewed in today’s post) that suggests that the aggregated form of the Parkinson’s associated protein alpha synuclein may be killing cells via the over activation of an enzyme called PARP. PARP over activation leads to increased levels of a protein called PAR, which firstly causes cells to die by the parthanatos cell death pathway, but also accelerates alpha synuclein aggregation by causing futher aggregation – resulting in a feed-forward cycle.
They have also found that treating models of Parkinson’s with PARP inhibitors reduces cell death and the accumulation of alpha synuclein, providing support for the re-purposing of this class of drugs use in Parkinson’s. As mentioned above, this alternative use of these drugs may be initially hampered by the inability of the compounds to enter the brain, but the results of the Dawson’s tireless efforts have provided yet another potential therapy for Parkinson’s and we can now expect a great deal of follow up work focused on this approach.
I don’t like giving opinions on this website, but I find this new report on PARP inhibition in models of Parkinson’s to be hugely exciting. The culmination of a long sequence of dedicated research, making a fascinating and easy story to tell. And it will be very interesting to see how this situation develops.
You can expect to hear more on the SoPD website regarding this topic as things progress.
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from Helix

These articles are awesome and give those of us with this disease, a glimmer of hope. I am 57 years old, a board certified residency trained emergency medicine physician.I was diagnosed approx 10 years ago and can no longer practice clinically. The clock is ticking … if you were me, which study would you try to get into? THX Steve
LikeLike
Hi Steve,
Thanks for your comment – glad you like the material. I get these kinds of questions a lot, and you will hopefully understand that I am very reluctant to give advice on such matters. This is for two reasons:
A. My background is that of a research scientist (not a clinician), and
B. Even if I was a clinician, it would be unethical for me to offer advice being unaware of the personal medical history/circumstances in each case.
At the same time I do not like to leave people with no information. So my approach to dealing with these questions is to explain what I would do if I were personally considering this matter. This way, I am providing information not advice, and the actions that you or others take based on that information would be your own responsibility. I hope you understand my position.
If I was diagnosed with PD and wondering which study to get involved with, I would firstly try to learn as much as I can about myself. This process would probably start with genotyping (to determine if I have a particular genetic risk factor) followed by bloods and any other assessments I could get. And this would solely guide my decision making process with regards to which studies to get involved with. For example, if I discovered in that effort that I have a GBA or a LRRK2 variant I would be currently looking at studies such as the Sanofi Genzyme (https://scienceofparkinsons.com/2018/01/10/gsls/) or the Denali (https://scienceofparkinsons.com/2018/07/25/lrrk-2/) studies, respectively.
I hope this helps.
Kind regards,
Simon
LikeLiked by 1 person
Steve, I’m a retired cell biologist 2 years into idiopathic PD. Early on I came across vitamin D3. It is a super-promoter influencing 10% of our genome, and enhances defences to oxidative stress, cytoplasmic calcium imbalance, and inflammation. My self-designed cocktail of endogenous agents is similar to Prof Wahl’s protocol for MS. High serum 25(OH)D3 is a major component.
As I see it, D3 trials for PD have been too short and over-cautious on dose.
Peter
LikeLike
And to get back on topic:
https://www.ncbi.nlm.nih.gov/pubmed/27561793
LikeLike
and:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2494848/
LikeLike
Hi Peter – agree with you re D3 – read an ebook on high dose years ago so – did 125K IU’s a day for over a year – went down to 50 for the next 6 months and now take 10K daily as maintenance – I too have a long list that was started after reading Dr. Wahls “Minding My Mitochondria” book – many more additions and changes since starting in 2012 or 13 – so – yes like many natural substances and or supplements – trials are woefully low in dosing – K2 dosing with high dose D3 per 10K IU’s is important too but all studies seem to conclude that single agents in low dose do nothing and should be avoided! D3 used to be available in 25 and 50mg doses OTC – that’s 100 and 200K IU’s respectively – why were concerned when someone gets over the artificially low RDA is beyond me – my level has been over 400 for years and my Parkinson Plus syndrome from which I graduated hospice with after 18 months having not died almost 6 years ago is stable or improving! Thanks for mentioning it!
LikeLike
Awesome! Thanks for the great description and for the info about the Dawsons. Nice to have faces to associate with brilliant work. Seems you were right that all the emphasis on alpha syn was somewhat misplaced.
LikeLike
Hi Diana,
Hope all is well – glad you liked the post. I found this was a really interesting topic, with intriguing potential for clinic application. Not sure the emphasis on alpha syn is completely misplaced, as this research suggests it is part of a feed-forward loop, but it will be interesting to see if PARP inhibitors can have an impact on PD in the clinic.
Kind regards,
Simon
LikeLike
The Linked Clinical Trials program has considered repurposing PARP inhibitors in PD for a few years, but enthusiasm has been dampened by fears of toxicity. The new data are encouraging, and perhaps lower doses of PARP inhibitors can be used with less risk, and still chances of benefit. You can read more about these ideas in Brundin, P. & Wyse, R. (2018) Cancer enzyme affects Parkinson’s disease. Science (New York, NY), 362, 521–522.
LikeLiked by 1 person
I was wondering if perhaps a *temporary* application of a PARP inhibitor could be used to break the cycle in which PAR causes greater alpha synuclein toxicity, resulting in more damage and thus more PAR production, etc. This might be done in parallel with the application of anti-inflammatories like baicalin, curcumin and EGCg, plus mitochondrial support such as ubiquinol, to break the feedback loop that goes through innate autoimmunity. I have an impression (well, a guess) that calming feedback loops below a certain threshold through the use of multiple treatment modalities might be the key to slowing or stopping PD progression.
LikeLike
Found your article (linked below), in which I think anyone reading this post would be interested. While my understanding is limited, it seems to be suggesting a combination of therapies to simultaneously and separately address both the PARP feedback loop (using a PARP inhibitor) and microglia-based autoimmunity (exenatide). I personally would place my faith more in such a multi-pronged approach than in the use of any single therapy.
http://www.sciencemagazinedigital.org/sciencemagazine/02_november_2018/MobilePagedReplica.action?pm=2&folio=521#pg25
LikeLike
Hi Lou T.,
Thanks very much for the link. You are right – it should be of interest to all of us.
Regarding your reference to “microglia-based autoimmunity (exenatide)”, here is a link that should provide some relevant information.
https://www.alzforum.org/news/research-news/does-taming-killer-astrocytes-spare-neurons-parkinsons-disease
Jeff
LikeLiked by 1 person
Thanks for that, Jeff. Prior to reading Dr. Brundin’s article, I had not been aware that astrocytes played a role in mediating the response of microglia to alpha-synuclein. I had thought that the cytokines generated by microglia (e.g., TNF-alpha) when their receptors (e.g., TLR4 and TLR2) were activated by alpha-synuclein and neuromelanin, directly triggered apoptosis (via a process that depends upon caspase-8) in neurons, including formerly-healthy “bystander neurons,” which then become damaged and release their own alpha-synuclein (and neuromelanin), starting the cycle all over again.
But the article you’ve linked indicates that the cytokines actually (or also?) incite astrocytes to transform into attack dogs that then attack the neurons. So I’m still a little confused regarding how astrocytes fit in, and whether or not there is a direct effect of cytokines on neurons, in addition to the indirect one mediated by astrocytes.
But anyway, if, as your linked article says, exenatide prevents astrocyte activation by inhibiting microglia, then we’re still talking about inhibiting microglia, with any astrocyte responses downstream from that. There are a number of things that inhibit microglia activation – Longvida curcumin, baicalin, EGCg, to name three. So, I’m wondering what is special about exenatide as opposed to these already-available OTC supplements. I would *think* that anything that inhibits microglial activation would also inhibit the downstream activation of astrocytes by cytokines.
So, my understanding of this is very incomplete, and in particular I’m unclear on how this treatment offers advantages over the substances just named above (curcumin, baicalin and EGCg).
LikeLike
That’s a good question (supplement vs drug). I had a look at the comment section of Simon’s earlier post on Exenatide and NLY01 (13 June 2018) and that question didn’t come up.
LikeLike
That’s possibly because I haven’t gotten around to reading that article yet. 🙂 I discovered this blog kind of late and so I’m gradually working my way backwards through the posts. Unfortunately, just about everything is essential reading, and it’s extremely dense material, so this is taking a while.
The research community is somewhat contaminated by drug money, but not nearly to the extent of clinical practice. In my very humble opinion. I mean, researchers are genuinely interested in anything that gets a result, but what gets funded seems to be largely drug product oriented. Especially when we’re talking about clinical trials.
Exenatide is an existing patented product that already has gone through safety and other human testing, so you can see how finding other possible uses for it as a treatment for PD could be enormously profitable. Whereas, curcumin, baicalin, EGCg, aren’t going to pay for anyone’s mortgage (or new yacht).
But just because the focus is very financially oriented does not mean that there won’t be interesting information and products. As patients, we need to keep asking questions about what these research findings might reveal regarding other substances for which there is little financial incentive to perform testing.
LikeLike
Hi Lou,
Thanks for all your comments. Much appreciated. Some of these comment sections are taking on a life of their own.
Exenatide is actually 12 months out of patent (it expired October 2017), but the nature of you statement still stands. I don’t think the research community is necessarily contaminated by industry money, but if industry being a new compound and says to researchers “we’ll fund you to investigate this”, few researchers are in a position to say “No thanks”. Their departments are all encouraging them to have closer ties with industry, and most scientists want to be seen to be team players in their departments. Plus industry have done away with their own research departments, so it is a necessary alliance unfortunately.
I do like the idea of the patient community continuing to ask questions. That kind of engagement was one of the early goals of this website.
Keep asking questions Lou!
Simon
LikeLiked by 1 person
Of course, the control of funds by corporations with a financial interest is part of the world in which we live. Researchers cannot be blamed for that fact, and there’s useful work to be done, even under such strictures. But patients do have to remain aware of the ways in which their interests may differ from those of the corporations who sponsor much of PD research, so that available treatments not coincident with corporate profits are not overlooked.
I remember you said in an earlier article regarding exenatide that “this drug is heavily protected patent-wise (there are one hundred and forty-three patent family members in twenty-one different countries for Exenatide! Three of these patents expire this year, four more in 2018 and 2020).” So perhaps there is still some profit to be made from finding new uses for that drug.
And also, I’ve noticed that not all drugs that go out of patent automatically revert to much-lower prices. I’m thinking of carbidopa, which seems to cost many times the price of sinemet that has an equivalent amount of carbidopa. I don’t know how they are able to keep prices so high for a generic drug with a manufacturing cost that is quite evidently a tiny fraction of its street price, but surely there must be some form of manipulation involved? This is something I’ve really wanted to understand, but still do not.
Still, it is a great thing that research into exenatide is occurring, and that it has yielded some promising results. I am not a purist in that regard, and neither do I believe that researchers should be. We live in the world that exists, not the world as we’d like it to be.
LikeLike
You’ll notice, though, that I *did* raise that question in this later article on Exenatide:
LikeLike
Mousing around just now, I found this very informative article that helps to answer my questions regarding how effective exclusivity can be maintained beyond the point that a generic version of a drug becomes legally possible.
https://www.raps.org/regulatory-focus™/news-articles/2016/8/patents-vs-market-exclusivity-why-does-it-take-so-long-to-bring-generics-to-market
“Take, for example, AstraZeneca’s diabetes treatment Bydureon (exenatide) and its patents and term of exclusivity as provided by FDA’s Orange Book (the Bible of pharmaceutical patent information). AstraZeneca has 18 patents covering the product, two of which don’t expire until 2026. However, the product’s market exclusivity expires in September 2018, which begs the question: Why does a drug have patents that do not expire until eight years after a generic could potentially come to market?
“Sherkow explains that this example is actually a typical case, noting: ‘Short answer here: this is the way generic entry is structured. For ANDAs [Abbreviated New Drug Applications – LT] to be approved, they need to certify that they don’t infringe any patents listed in the Orange Book … which is one of the reasons they’ll add these patents to discourage generic companies from filing ANDAs in the first place,’ particularly because of the expense of the litigation.
“Elaine Blais, head of the litigation department at Goodwin Procter’s Boston office, also told Focus that under Hatch-Waxman, which is the Act outlining the process by which an ANDA can be filed, an ANDA is often subject to an automatic 30-month stay of approval when a brand name drug company files a patent infringement suit.”
LikeLike
And then there’s just the outright payoff approach, which under Obama apparently declined, but now under Trump will it go back to business as usual?
https://www.raps.org/regulatory-focus™/news-articles/2016/1/ftc-pay-for-delay-deals-decrease-significantly-after-supreme-court-ruling
LikeLike
One truly does get the feeling that researchers are closing in on a detailed understanding of how PD happens. It’s a very complicated picture, but you’ve made it surprisingly clear.
I’m wondering, though, whether inhibiting PARP is perhaps just one part of what’s required to slow down PD progression.
Where does damping down microglia-based inflammation fit into this scheme? My understanding is that when dopamine neurons die, they spill their guts (including neuromelanin and aggregated alpha-synuclein) into the inter-cellular space, and that activates microglia, which generate cytokines, causing more neurons to die, starting the autoimmune feedback loop all over again.
So, once that autoimmune feedback gets started, maybe inhibiting PARP could be one part of damping it down (by reducing alpha-synuclein aggregation, and cell death from the specific cause of Parthanatos), but there would still be the dumping of neuromelanin and monomeric alpha synuclein, which I believe would still tend to cause microglial activation, even without aggregated AS as one part of that mix.
According to this article, microglial cytokine release operates via an apoptotic pathway that does not require PARP:
https://www.ncbi.nlm.nih.gov/pubmed/23760205
“TNF-induced necroptosis and the PARP pathway represent distinct and independent routes to programmed necrosis…inhibition of the PARP pathway does not protect against TNF-induced necroptosis…”
More generally, there’s a difference between protecting healthy neurons (e.g., by inhibiting PARP) from the effects of a new assault by MPTP or whatever, on the one hand, and damping down the effects of an established, neuron-destroying PD process (e.g., innate autoimmunity), on the other.
I’m wondering also about the cancer risk of inhibiting PARP. In PD, PARP is overproducing PAR for a *reason*, right? What *is* that reason? Is there extensive DNA damage that PARP is trying to repair? If so, will inhibiting PARP allow that damage to persist, and allow the neurons that contain that damaged DNA to reproduce? Or is there another process that will ensure the apoptosis of already-damaged cells before they can reproduce and become tumors (for example, p53, if that can still work without PARP to signal that there is damage)?
Now, if the DNA damage is *itself* due to the Parthanatos process, then maybe inhibiting PARP *temporarily* could break that feedback of DNA damage causing too much PARP/PAR causing Parthanatos causing DNA damage, etc. And then, after that cycle was damped down, you could remove the PARP inhibitor and let PARP get back to doing its normal repairs at a normal level…?
But if the DNA damage is from some other source (e.g., innate autoimmunity), then wouldn’t one would have to remove that problem in order to not need the PARP inhibitor on a continuing basis?
These may be questions that do not presently have answers. Or, maybe they do, and I will be fortunate to read them here!
Anyway, this seems like an amazing advance in our understanding the illness, and I am so looking forward to the hearing about the tests of PARP inhibitors in human trials.
LikeLike
Hi Lou,
Great comment. Lots of questions.
The new study suggests that rather low doses of the PARP inhibitors could be used for PD, much lower than the doses used in oncology. This means that over active PARP and high levels of PAR could be quelled while still leaving natural DNA repair processes to do their thing (hopefully). This would also reduce potential side effects. The issue at the moment, however, is that pesky blood brain barrier, but there are ways to deal with that which are being explored.
Kind regards,
Simon
LikeLiked by 1 person
Just wanted to add a note that quercetin and fisetin have been identified as PARP-1 inhibitors, with a very low risk profile and of course OTC.
https://www.ncbi.nlm.nih.gov/pubmed/17884996
LikeLike
Simon,
Thank you for another informative post. You wrote : “most of the available PARP inhibitors have very poor penetration of the brain. They all have a lot of trouble getting across the blood-brain-barrier – the protective membrane surrounding our brains. ” John Hopkins University looked at the repurposed medication Lexiscan that can modulate permeability of the BBB: https://www.jhunewsletter.com/article/2016/04/drug-repurposed-to-activate-endothelial-cells
Would not it be interesting to try this approach with PARP inhibitors? For that matter it might be a good idea to try it in the Prothena clinical trial where the published report stated that CSF concentration of ASN specific antibodies was only .2 or .3%?
Thanks,
Felix
LikeLiked by 1 person
Hi Felix,
Thanks for the really interesting comment. It could be worth further investigation. Will look into it.
Kind regards,
Simon
LikeLiked by 1 person
In the graphic titled ‘DNA Damage’ (credited to Sierra Oncology), are the labels for ‘endogenous’ and ‘exogenous’ stressors swapped? Exogenous means from external sources – radiation would be an external source of stress leading to DNA damage, but is under the label ‘endogenous’.
LikeLike
Hi Rhyo,
Thanks for pointing out the error – you are correct and I completely missed it (Ha!). I will try and find an alternative image to replace it.
Kind regards,
Simon
LikeLike
All excellent comments – and reply’s – wondering a few things – 1) no one has discussed the difference between senescent vs dead (necrotic or whatever) cells – I do believe that senescent cells can be revived given the right CPR (cellular processing resuscitation) – no current technology can identify the total number (millions) of cells that are either in the process of dying or have died and those that are senescent and could possibly be revived! 2) Why are there cells that are not removed via apoptosis – what makes the immune system or the apoptotic mechanisms malfunction such that cells remain and are not removed? 3) citizens united and corporate lobbying with unlimited funds create the atmosphere that what they want to generate more capital is what they are working on and will go to any lengths to accomplish that which is not limited to fake peer review by ghost writers and manipulated data like the recent FDA review – the system is broken! 4) Natural substances and anti inflammatory diet seem to be sorely neglected in research for the reason in #3 – Diet Against Disease linked below has postings re dysbiosis barrier disruption and autoimmune activation with alphasynuclean generation – reversing the process that started it instead of trying to intervene with drugs somewhere in the middle to me seems to make more sense but that would not be considered by research would it? Just some thoughts needing an accounting!
LikeLike