
|
At 23:30 on the 3rd August 2017, the results of a phase II clinical trial investigating the use of a Glucagon-like peptide-1 receptor (GLP-1R) agonist called Exenatide (Bydureon) in Parkinson’s were published the Lancet journal website. The findings of the study were very interesting. And after years of failed trials, the Parkinson’s community finally had a drug that appeared to be ‘doing something’. Naturally these results got many in the Parkinson’s community very excited. Over the last couple of weeks, further research related to this topic has been published. In today’s post we will review some of this new research and ask some important questions regarding how to move forward with these results. |

In 2012, the Golden Goose Award was awarded to Dr John Eng, an endocrinologist from the Bronx VA Hospital.

Dr John Eng. Source: Health.USnews
The Award was created in 2012 to celebrate researchers whose seemingly odd or obscure federally funded research turned out to have a significant and positive impact on society.
And despite the name, it is a very serious award – past Nobel prize winners (such as Roger Tsien, David H. Hubel, and Torsten N. Wiesel) are among the awardees.
This week a research report was published in the journal Nature Medicine that expanded on the work of Dr Eng (some 25 years after his big discovery).
And it could be very important to the Parkinson’s community.
Sounds intriguing. What did Dr Eng do?
In the 1980’s, Dr Eng became really interested in some studies (such as this one) that described the effects that certain types of venom had on cells in the pancreas.
The pancreas is the organ that produces the chemical insulin which is critical for maintaining normal glucose levels in our bodies. Having worked with diabetic people who do not produce enough insulin, Dr Eng started wondering if venom may contain chemicals that could help people with diabetes. But rather than injecting diabetic people with venom, he started looking at all the chemicals that make up the venom from different poisonous creatures.
Venom looks like great fun. Source: Cen
What did he find?
This charming creature is a Gila monster.

The Gila monster. Source: Californiaherps
Cute huh?
Some interesting facts about the Gila (pronounced ‘Hila’) monster:
- They are named after the Gila River Basin of New Mexico and Arizona (where these lizards are found)
- They are protected by State law
- They are venomous, but very sluggish creatures
- They spend 95 percent of their time underground in burrows.
In 1992, Dr Eng identified the two proteins that he had isolated from the venom of the Gila monster. One of them was called exendin-4 and it bore a striking similarity -structurally and functionally – to a human protein, called glucagon like peptide-1 (GLP-1).
What is GLP-1?
Insulin instructs cells to take in and use glucose from the blood. This has the effect of lowering blood sugar. The hormone Glucagon has the opposite effect – it tells the body to release glucose into the blood to raise sugar levels.
GLP-1 is a hormone that stimulates insulin production while blocking glucagon release.

The function of GLP-1. Source: Wikipedia
Unfortunately, naturally produced GLP-1 in your body is rapidly deactivated by a circulating enzyme called dipeptidyl peptidase IV. Exendin-4, however, was found to be resistant to this deactivation, meaning that could last longer in the body stimulating insulin production and blocking glucagon release. Dr Eng quickly realised that there was enormous medicinal potential for exendin-4 as a drug for people with diabetes. He patented the idea and soon afterwards a biotech company called Amylin Pharmaceuticals to begin the work of turning exendin-4 into a drug for diabetes.
That drug was eventually called Exenatide.
In April 2005, Byetta (the commercial name for Exenatide) was approved by the FDA for clinical use in the treatment of Type 2 diabetes. On the 27th January 2012, the FDA gave approval for a new formulation of Exenatide called Bydureon, as the first weekly treatment for Type 2 diabetes. In July of 2012, Bristol-Myers Squibb announced it would acquire Amylin Pharmaceuticals for $5.3 billion, and one year later AstraZeneca purchased the Bristol-Myers Squibb share of the diabetes joint venture.
Interesting, but what does any of this have to do with Parkinson’s?
In 2008, this report was published:

Title: Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease.
Authors: Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, Kortesmaa J, Mercer A, Nielsen E, Rönnholm H, Wikström L.
Journal: J Neurosci Res. 2008 Feb 1;86(2):326-38.
PMID: 17803225
In this study, Exendin-4 (the protein very similar to exenatide) was tested in a rat model of Parkinson’s. Five weeks after giving the neurotoxin (6-hydroxydopamine) to the rats, the investigators began treating the animals with exendin-4 over a 3 week period. Despite the delay in starting the treatment, the researchers found behavioural improvements and a reduction in the number of dying dopamine neurons.
And this first result was followed a couple of months later by a similar report with a very similar set of results:

Title: Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease.
Authors: Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS.
Journal: J Neuroinflammation. 2008 May 21;5:19. doi: 10.1186/1742-2094-5-19.
PMID: 18492290 (This study is OPEN ACCESS if you would like to read it)
The scientists in this study tested exendin-4 on two different rodent models of Parkinson’s (6-hydroxydopamine and lipopolysaccaride), and they found similar results to the previous study. The drug was given 1 week after the animals developed the motor features, but the investigators still reported positive effects on both motor performance and the survival of dopamine neurons.
And the following year, in 2009, two more research reports were published suggesting that exendin-4 was having positive effects in models of Parkinson’s (Click here and here to read those reports).
This was a lot of positive results for this little protein.
How is Exendin-4/Exenatide having this positive effect?
Exendin-4 and exenatide are both GLP-1 receptor agonists.
What does that mean?
On the surface of cells there are small proteins called receptors, which act like switches for certain biological processes. Receptors will wait for another protein to come along and activate them or alternatively block them.
The proteins that activate the receptors are called agonists, while the blockers are called antagonists.

Agonist vs antagonist. Source: Psychonautwiki
Exendin-4 and exenatide are agonists, so they activate the GLP-1 receptor.
Activation of the GLP-1 receptor by a GLP-1 receptor agonist like exendin-4 or exenatide results in the activation of many different biological pathways within a cell:

The GLP-1 signalling pathway. Source: Sciencedirect
Of particular importance is that GLP-1 receptor activation inhibits cell death pathways, reduces inflammation, reduces oxidative stress, and increases neurotransmitter release. All pretty positive stuff really. For a recent and very good OPEN ACCESS review of the GLP-1-related Parkinson’s research field, click here.
And all of these research reports with positive results led to and supported the idea of clinically testing exenatide in people with Parkinson’s.
What happened in the first clinical trial?
The first clinical trial of exenatide in Parkinson’s was a phase I trial to determine if the drug was safe to use in people with Parkinson’s. The results of the trial were published in 2013:

Title: Exenatide and the treatment of patients with Parkinson’s disease.
Authors: Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T.
Journal: J Clin Invest. 2013 Jun;123(6):2730-6.
PMID: 23728174 (This study is OPEN ACCESS if you would like to read it)
The researchers gave exenatide (the Byetta formulation which is injected twice per day) to a group of 21 people with moderate Parkinson’s and evaluated their progress over a 14 month period. They compared those participants to 24 additional subjects with Parkinson’s who acted as control (they received no treatment). Exenatide was well tolerated by the participants, although there was some weight loss reported among many of the subjects (one subject could not complete the study due to weight loss).
Importantly, the exenatide-treated subjects demonstrated improvements in their Parkinson’s movement symptoms (as measured by the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (or MDS-UPDRS)), while the control patients continued to decline.
Interestingly, in a two year follow up study – which was conducted 12 months after the subjects stopped receiving exenatide – the researchers found that participants previously exposed to exenatide demonstrated a significant improvement (based on a blind assessment) in their motor features when compared to the control subjects involved in the study.
It is important to remember, however, that this trial was an ‘open-label study’ – that is to say, the participants knew that they were receiving the exenatide treatment so there is the possibility of a placebo effect explaining the improvements. And this necessitated the testing of the efficacy of exenatide in a phase II double blind clinical trial.
And the results of that trial were published last August (2017):

Title: Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial
Authors: Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, Li Y, Aviles-Olmos I, Warner TT, Limousin P, Lees AJ, Greig NH, Tebbs S, Foltynie T
Journal: Lancet 2017 Aug 3. pii: S0140-6736(17)31585-4.
PMID: 28781108
In the study, the investigators recruited 62 people with Parkinson’s (average time since diagnosis was approximately 6 years) and they randomly assigned them to one of two groups, exenatide (the Bydureon formulation which is injected once per week) or placebo (32 and 30 people, respectively). The treatment was given for 48 weeks (in addition to their usual medication) and then the participants were followed for another 12-weeks without exenatide (or placebo) in a ‘washout period’.
It is important to remember that in this trial everyone was blind. Both the investigators and the participants. This is referred to as a double-blind clinical trial and is considered the gold standard for testing the efficacy of a new drug.
The researchers found a statistically significant difference in the motor scores of the exenatide-treated group verses the placebo group (p=0·0318). As the placebo group continued to have an increasing (worsening) motor score over time, the exenatide-treated group demonstrated improvements, which remarkably remained after the treatment had been stopped for 3 months (weeks 48-60 on the graph below).
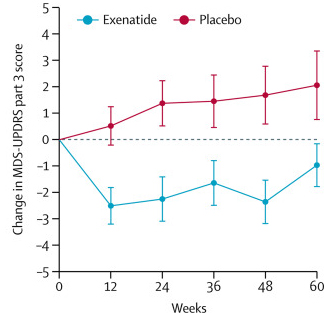
Reduction in motor scores in Exenatide group. Source: Lancet
Brain imaging (DaTscan) also suggested a trend towards reduced rate of decline in the exenatide-treated group when compared with the placebo group. Interestingly, the researchers found no significant differences between the exenatide and placebo groups in scores of cognitive ability or depression – suggesting that the positive effect of exenatide may be specific to the dopamine or motor regions of the brain.
Given that these results were coming from a randomised, double-blind clinical trial, the Parkinson’s community got very excited about them (Click here to read a previous SoPD post about this particular trial).
This is very positive stuff. What has been published more recently?
So very recently, the researchers who conducted the Phase II exenatide trial published some follow up results which looked deeper into the data and found some interesting trends:

Title: What Effects Might Exenatide have on Non-Motor Symptoms in Parkinson’s Disease: A Post Hoc Analysis
Authors: Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Skene S, Chowdhury K, Aviles-Olmos I, Limousin P, Foltynie T
Journal: Journal of Parkinson’s Disease, 2018, Pre-press, pp. 1-12.
PMID: N/A
In this study, the investigators collected all of the data that had been collected during the exenatide clinical trial, and they went through the painstaking process of trying to find interesting trends in the data that were not large enough to be picked up in the initial analysis.
And as they analysed all of the data, the researchers noticed something interesting.
Compared to the placebo-treated group, participants treated with exenatide appeared to have improvements in individual domains assessing mood and depression. While the trends were not always significant, they were apparent across all observer-rated outcome measures that were collected after 48 weeks.
These changes were not associated with changes in motor severity or other factors, which the investigator proposed may suggest that exenatide could be having an independent effect on mood.
BUT, we have to be very careful in how we interpret these results.
And the researchers have stressed throughout the report that these new results are based on a post hoc analysis, which in no way should be interpreted as evidence of efficacy (irrespective of any statistical threshold).
This type of analysis is only conducted for hypothesis generating purposes (especially on such a small sample of participants – just 32 people treated with exenatide).
Having said that, the results of this new analysis may also be very useful in the planning of outcome measures in the next exenatide (or other future GLP1 agonist) clinical trials.
Very interesting. So summing up, what does it all mean?
Not just yet.
We aren’t finished.
There is more to discuss.
Que?
Earlier this week a research report was published on the website of the journal Nature Medicine, which may help to explain how exenatide could be having an effect on Parkinson’s… and it may also provide another GLP-1 receptor agonist to add to the arsenal of new treatments for PD.
Here is the report:

Title: Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease
Authors: Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang SU, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS
Journal: Nature Medicine, 2018, 11 June. [Epub ahead of print]
PMID: 29892066
(Before we start here, I just want to say: This report is a beast! It is massive – so much work has been done! Congrats to the investigators on an impressive volume of work).
In this study, the researchers were investigating the use of a drug called NLY01 in models of Parkinson’s. NLY01 is new GLP-1 receptor agonist being developed by a company called Neuraly Inc, which is also developing novel brain penetrant c-Abl inhibitors for Parkinson’s (Click here to read more about this).

The investigators started their analysis by treating normal mice and primates with the drug.
NLY01 had a very impressive half-life of 38 hours in mice and 88 hours (in primates). This is impressive compared to Exenatide (Byetta) which has a half-life of 2.5 hours in primates. The half-life of a drug is the time required for the concentration of the drug to decrease by half in the body.
NLY01 also did not increase the risk of hypoglycemia (or low blood sugar) when administered in the animals. And it reached the brain very efficiently – having no issues with the blood brain barrier (the protective membrane surrounding the brain).
Next, the researchers tested the drug on two different mouse models of Parkinson’s:
- the alpha synuclein preformed fibril model
- the hA53T genetically engineered mouse
What are alpha synuclein preformed fibrils?
As regular readers will be aware alpha synuclein is considered to be one of the main trouble makers in Parkinson’s. It may sound like a distant galaxy, but it is an extremely abundant protein in our brains – making up about 1% of all the proteins floating around in each neuron.
When alpha synuclein is produced by a cell, it normally referred as a ‘natively unfolded protein’, in that is does not really have a defined structure. Alone, it will look like this:
Alpha synuclein. Source: Wikipedia
By itself, alpha synuclein is considered a monomer, or a single molecule that can bind to other molecules. When it does bind to other alpha synuclein proteins, they form an oligomer (a collection of a certain number of monomers in a specific structure). In Parkinson’s, alpha synuclein also aggregates to form what are called ‘fibrils’.

Microscopic images of Alpha Synuclein (AS) monomers, oligomers and fibrils. Source: Brain
And over the last few years research labs have been taking alpha synuclein and turning them into fibrils, and then using those ‘preformed fibrils’ in models of Parkinson’s. For example:

Title: Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice.
Authors: Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM.
Journal: Science. 2012 Nov 16;338(6109):949-53.
PMID: 23161999 (This report is OPEN ACCESS if you would like to read it)
In this study, when the researchers injected preformed fibrils of alpha synuclein into the mice, they started to observed alpha synuclein protein aggregation in the brain approximately one month later, and the mice started to exhibit motor problems 3-6 months later.
In the NLY01 study, the researchers injected their preformed fibrils into mice and they waited one month before initiating treatment of the mice with either NLY01 or a placebo treatment. They treated the mice for twice per week for 5 months.
The results found that NLY01 treatment significantly reduced the amount of alpha synuclein aggregating and also reduced the loss of dopamine neurons. The drug also rescued the motor deficits across a range of behaviour tests.
Next the researchers tested NLY01 on the hA53T genetically engineered mouse.
And what is the hA53T genetically engineered mouse?
There is a region of your DNA that provides the instructions for making alpha syncuclein. That region of DNA is called SNCA. And there are several genetic variations (or mutations) inside of SNCA that are associated with an increased risk of developing Parkinson’s.
A53T is the name of one of those genetic variations.
As you can see in the image below, A53T lies in the red (Amphipathic) region of SNCA along with several other genetic variants, such as A30P and E46K:
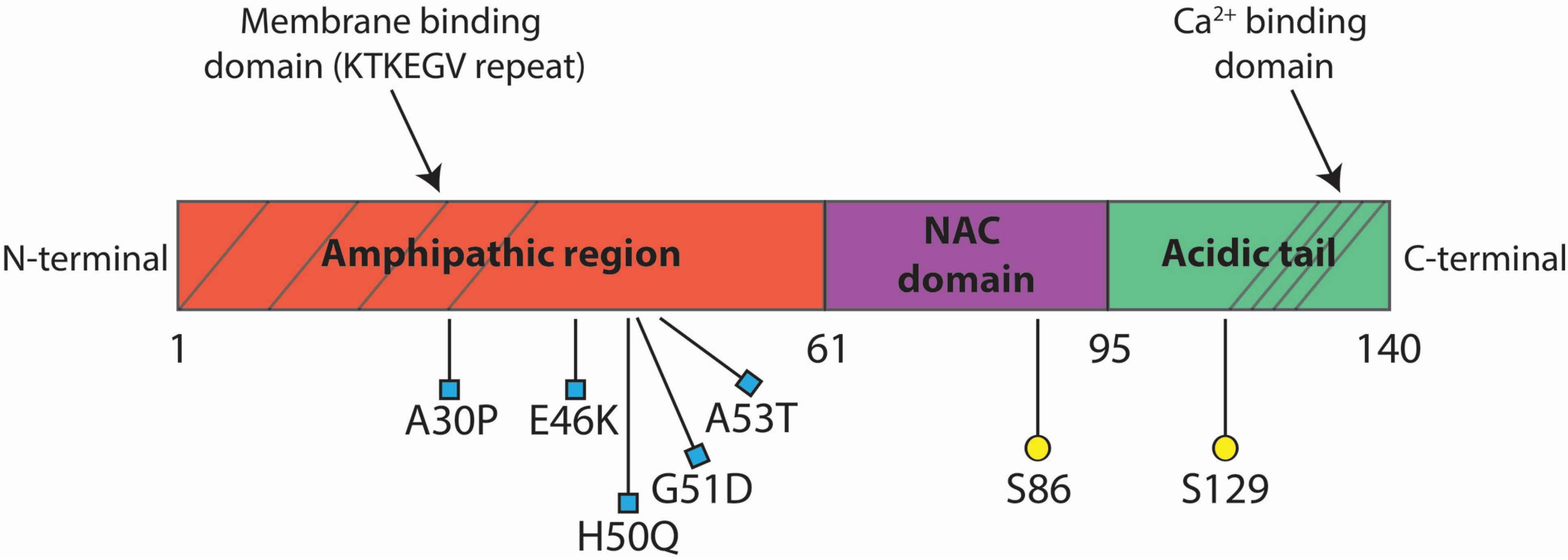
Mice have been genetically engineered to carry the SNCA gene with the A53T genetic mutation (Click here to read the original report). These mice exhibit hyperactivity and then start to display signs of alpha synuclein protein aggregation at about four to six months of age. They also pass away earlier than normal mice (12-14 months of age, compared to 20+ months for normal mice).
In the NLY01 study, the investigators started treating the A53T mice at 6 months of age with either NLY01 or a placebo control, and they reported that NLY01 significantly prolonged the lifespan of the A53T mice by over 3 months. In addition to this prolonged survival, the researchers also found a reduction in levels of aggregated alpha synuclein in the brains of the NLY01-treated A53T mice.

Source: PBS
Next, the researchers wanted to try and determine how NLY01 was having this effect (the ‘mechanism of action’), and they started by labelling GLP-1 receptors in cell cultures of difference types of brain cells (astrocytes, microglia and neurons) and then they looked to see where GLP-1 was most abundant.
Interestingly, they found that microglia cells have the highest levels of GLP-1 receptors.
Microglia are some of the helper cells in the brain. They act as the resident immune cells. When infection or damage occurs, the microglia become ‘activated’ and start cleaning up the area.
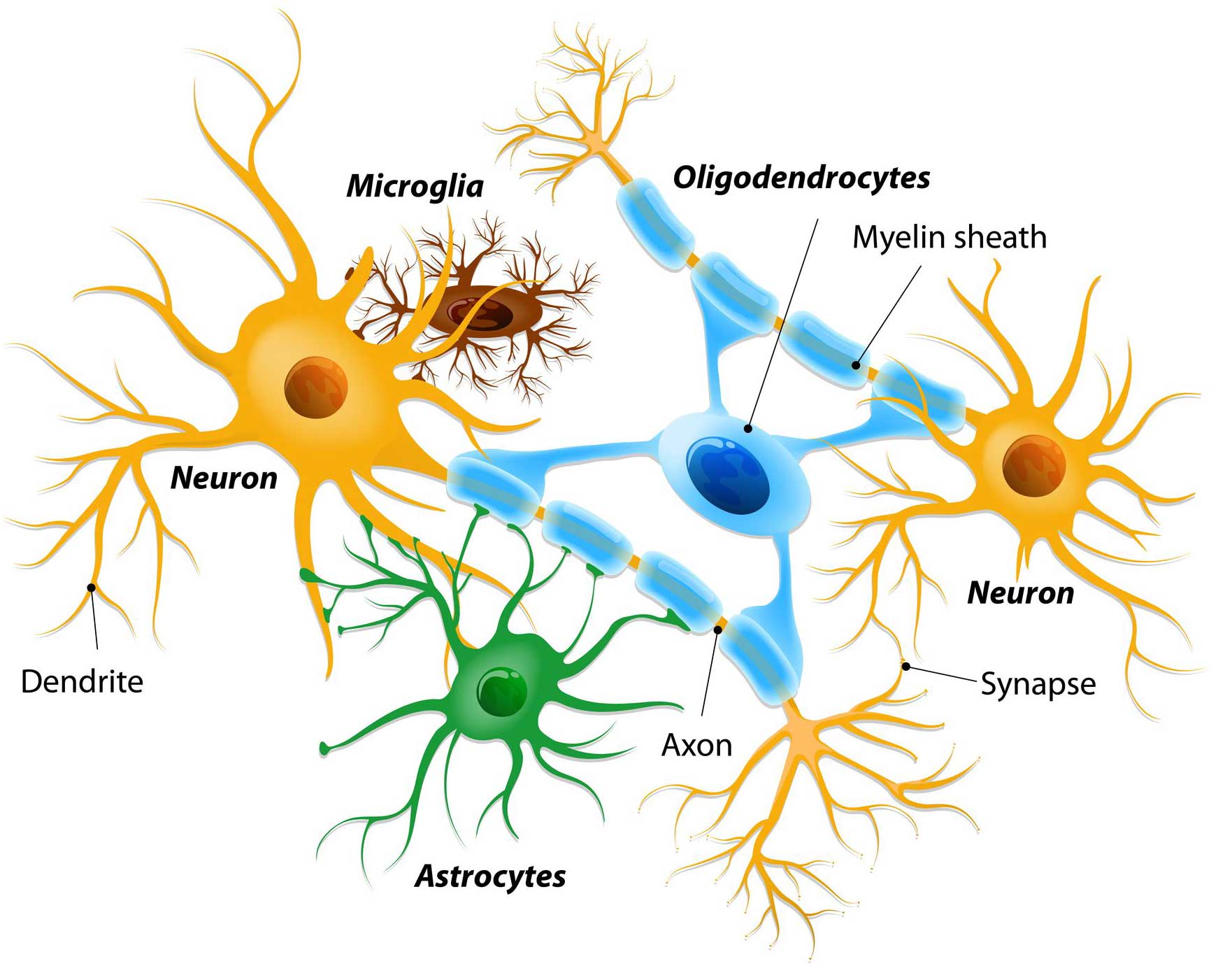
Different types of cells in the brain. Source: Dreamstime
Also of interest: the researcher reported that treating the cells with preformed fibrils of alpha synuclein decrease GLP-1 levels in neurons and increased GLP-1 levels almost two fold in microglia. And when they looked at postmortem tissue, the investigators found a 10x increase in the levels of GLP-1 in the substantia nigra of people who passed away with Parkinson’s (compared to healthy controls samples). The substantia nigra is the region of the brain where the dopamine neurons reside, and it is badly affected in Parkinson’s.
The finding that microglia have higher levels of GLP-1 protein led the researchers to test the idea that perhaps NLY01 was having its effect via the microglia cells rather than the neurons. So they treated cultures of just dopamine neurons with preformed fibrils of alpha synuclein and the investigators found that NLY01 treatment had no protective effect on the cells.
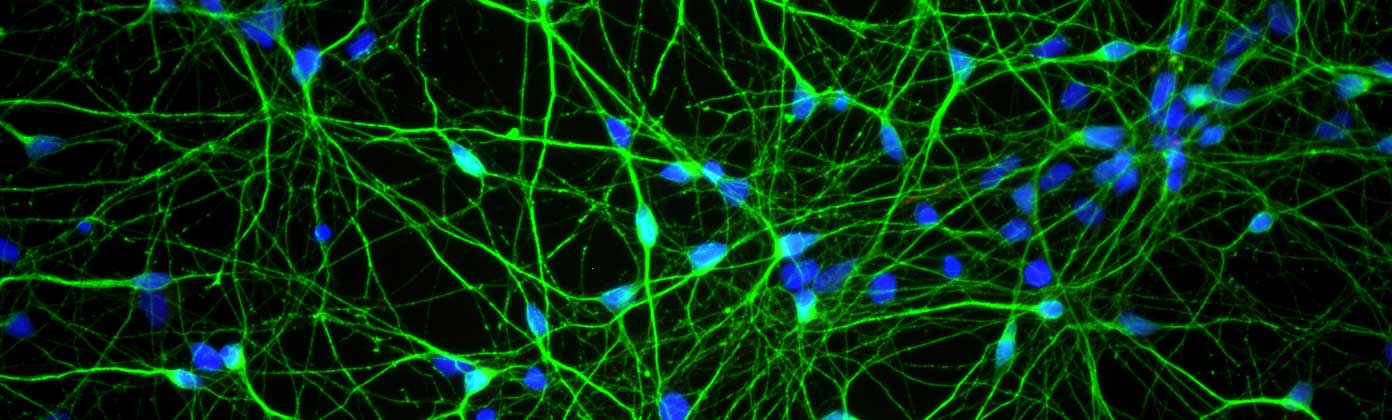
Dopamine neurons (green) in cell culture. Source: Axolbio
Previously the researchers conducting this study had demonstrated that activation of microglia can cause them to release all sorts of nasty, inflammatory chemicals. They do this to alert other microglia to a potential problem. But these chemicals can also cause astrocytes to shift from a neutral state to a highly reactive state (Click here to read more about this).

An astrocyte. Source: Wikipedia
Astrocytes, like microglia, are helper cells in the brain. They provide nutrients to neurons and make sure the environment surrounding the neurons is balanced and supportive. When astrocytes shift from a neutral state to a reactive state, however, it can be very bad news for any neurons nearby. And given this scenario, the researchers began to wonder whether the interactions between microglial cells and astrocytes could be involved in the neuroprotective effects of NLY01.
They decided to test this idea by firstly treating microglia with preformed fibrils of alpha synuclein. This got the microglia nice and activated, and the researchers then split the microglia in to two groups: one group was treated with NLY01 and the other group was not. Next the investigators collected the solutions from these two groups of microglia cultures, and they applied those solutions to two groups of astrocytes in cell culture. Astrocytes given the NLY01-treated microglial solution did not become reactive.
And subsequent experiments demonstrated that this reduction in reactive astrocytes was neuroprotective for dopamine neurons when they were grown in culture solution from the NLY01-treated activated microglia and reactive astrocytes.
I hope all of this makes a little bit of sense.
Long story short/the take home message:
- Preformed fibrils of alpha synuclein activate microglia
- Activated microglia spit out nasty, inflammatory chemicals
- Astrocytes exposed to those chemicals get reactive
- Reactive astrocytes are bad news for nearby neurons
And NLY01 limits the activated microglia from spitting out too much of the nasty, inflammatory chemicals, thus lowering the number of reactive astrocytes.
NLY01 appears to be having its effect via the microglial cells.
All clear?
Interestingly, when the researchers removed astrocytes from the equation by exposing the neurons to solution collected from activated microglia, there is some cell death in the neuronal population, but it is not as much as that seen in the cultures including reactive astrocyte solution.
Thus, reactive astrocytes seem to be critical to the neuronal cell death.
And when the researchers looked at the brains of the ‘preformed fibrils of alpha synuclein’ injected mice that had been treated with NLY01 for 5 months (which we discussed above), they found a reduced number of activated microglia.
These results led the researchers to conclude that “NLY01 exhibits protective effects primarily through microglial GLP1R“. And because of this specific mechanism, the investigators suggest that “NLY01 may have broad neuroprotective properties in a variety of neurodegenerative disorders and neurologic injuries“, beyond just Parkinson’s.
All of this data sounds fantastic. So why the negative title to this post?!? Exenatide doesn’t sound like a problem.
Ah, but it is.
In several ways.
(and if you are not interested in academic arguments, you can turn off here)
Let’s me ask you this: With all the hype and excitement in the Parkinson’s community surrounding Exenatide, how can we possibly conduct an un-biased phase III clinical trial?
And this is a very serious question.
Massive resources (millions of dollars and hundreds of people) are potentially going to be marshaled in a phase III clinical trial of this drug. Multiple research centres are going to be involved. It would be a major investment.
Source: Cancer.umn
But expectations are already extremely high (and websites like the SoPD are obviously not helping!).
We have been to this rodeo before (think of all the GDNF approaches to PD – they had high expectations). With such high expectations, the risk of a placebo response (that is, the experiencing of beneficial effect where no pharmacological cause is apparent) is a very real possibility. And the placebo effect is a major issue in Parkinson’s research (Click here to read a previous post on the placebo effect in PD).
Imagine conducting a massive (200+ participant) clinical trial and at the end of the 2 years period of the study finding that many participants in the placebo control group demonstrated a placebo response (that is, they had beneficial effects despite only taking the the inert treatment).
Thus, we return to the question: With all the hype and excitement in the Parkinson’s community surrounding Exenatide, how can we possibly conduct an un-biased phase III clinical trial?
Your thoughts on this matter in the comment section below would be greatly appreciated.
And then, if we do run “Exenatide III” and the results are positive, will we face the ‘Dilemma of success‘? That is, if we have a semi-neuroprotective drug that everyone wants to take, how will we develop future neuroprotective therapies for Parkinson’s?
As I have said before, “these are good problems to have“, but they do keep me awake at night!

Source: Trainright
So what does it all mean?
These are really interesting times for Parkinson’s research. And it is hard not to be excited – it is a very human response when there are some positive results after a long period of negative outcomes. We may be standing on the edge of a new age with regards to Parkinson’s, where sufferers may be provided with more than just treatments that cover up the symptoms.
But it is important for readers to understand that we are not there yet.
The results coming from the GLP-1 agonist research are very interesting and encouraging, but we must not read too much from them. The deep analysis of the Phase II clinical trial ‘suggest’ that exenatide may be having an effect that goes beyond the previously reported motor results, but all of these results need to be tested in a larger cohort and over a longer period of time. There is still so mush that we don’t know. For example, we have no idea what impact long term exenatide treatment has in Parkinson’s (diabetes yes, but PD may be different – we simply do not know).
One of the ‘long-term use’ concerns with the exenatide approach to Parkinson’s is the weight loss issue. A lot of people that take exenatide lose their appetite. And this was evident in the phase II exenatide study, in which at the 48 weeks time point, the exenatide group had lost an average of 2·6 kg, while the control group had lost only 0·6 kg. In the phase I trial one subject could not complete the study due to weight loss.
Thus, there is still a lot of work to be done, but the results are looking very encouraging.
ADDENDUM – 18/07/2018: Johns Hopkins announced today the launch of Neuraly Inc, with $36 million in financing that will be used to begin phase 1 clinical testing of their GLP-1 agonist NLY01 in people with Parkinson’s before the end of the year (Click here to read the press release).
Things are certainly starting to move quickly around here.
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from Businesswire

Great post!
LikeLiked by 3 people
Thanks Pamela! Glad you liked it
LikeLike
I’m currently in a 2-year study and it seems to drag on. I’m tired of it — bored of the UPDRS and the million questions they rattle off in rapid succession. I think they’re tired of it too. And they keep asking for multiple dat scans. Ugh!
If you get assigned to placebo it’s a restive state to be in with new news appearing every month. You soon feel trapped in an older less interesting study demonstrating the most minor effects if any.
It’s too bad they can’t shorten the time. Maybe the gold standard is too high. Give it a year then make it available. Let patients sign off on the risks. Time will tell the story. In reality two years is only telling a short term story.
LikeLike
Isn’t placebo response shorter in duration than real effects? But maybe not short enough for a “short” study, if Diana can forgive that wording.
LikeLike
Hi Dkdc,
Actually there are cases of placebo effect lasting 12+ months in clinical trials of Parkinson’s (see the mention about the Freed et at (2001) study on this post: https://scienceofparkinsons.com/2016/01/03/the-placebo-effect-and-parkinsons-disease/). I only wish we could bottle some of that stuff underlying the placebo effect and offer it to the Parkinson’s community.
Kind regards,
Simon
LikeLike
Hi Diana,
Well done for partaking in a clinical trial – it is a major commitment that I fully respect. I also appreciate the comment about the ‘dragging’ aspect of it. One of the biggest issues with the large trials is adherence and keeping participants enrolled, and I have often wondered how many of the dropouts in trials simply give up. Do you think it’s an engagement issue? Do you feel engaged?
Kind regards,
Simon
LikeLike
I wild love to see two modes/levels of your more then veluble (INNOVATIVE) PD posts. As usual this HI Q “sci-PD reviews” and all these brilliant questions regarding PD, but also a “not so sci” (short) mode of same post suitable for the wider public. Thank You!
LikeLike
Hi Radosla,
Glad you like the material, but I’m afraid there is currently barely enough time in the day for the SoPD let alone work and everything else! I fully appreciate that this website is a bit niche and that most people don’t want the biology lesson. In those cases, I usually point people towards the Parkinson’s UK medium page (https://medium.com/parkinsons-uk) or the Parkinson’s New Today website (https://parkinsonsnewstoday.com/). They provide a fantastic overview of what is happening in the world of Parkinson’s research in a less sciency-manner.
Ok, lunch break is over – I better get back to work,
Simon
LikeLiked by 1 person
Thanks, my satisfaction from your very informative replay is 99.99 %
LikeLike
I’m with Diana. Except, if it weren’t for those studies, how would we know what’s promising?
I’d like very much to see these drugs being tested on sufferers with RBD to see if they slow or stop progression to full scale Parkinsonian or non-Parkinsonian syndromes like MSA.
Seems to me we have a terrific opportunity by targeting this cohort.
Me? I’m not waiting for an official RBD diagnosis. I’ve awakened several times moving in the middle of nightmares over the past two decades (it isn’t frequent), but having been tested for Apnea and being found negative, I decided to do several things.
1) I’m already on a CCB, so I switched to isradipine, which BTW has fewer side effects than my old drug and is a bit more effective on my Arterial Hypertension, which is now around 120/75. No need to give vascular dementia any help, right? BTW, I’ve not had a single waking movement since I started. If it proves out not to be effective against PD, no harm, no foul.
2) Low dose selegiline. I was convinced by the research that it should be started by anyone over the age of 45 as a life-extension drug. So far, so good.
3) I put mannitol in my coffee. Why not? Helps keep me regular, doesn’t raise my already insulin-resistent (especially in the mornings) glucose levels. Helps keep my A1C around 5.
4) I take Metformin for pre-diabetes (yeah, I exercise and diet, too). But it seems like a wonder drug for it’s anti-cancer properties and is helpful in warding off Type 2 diabetes.
I have a whole regimen of life-extension supplements that have some good evidence of effectiveness, so, why not? Glycine could be protecting my brain stem (where RBD apparently starts), and probiotics might be protecting it as well by keeping some sort of prionic whatever from climbing the Vegus nerve into the brain stem.
My only problem with Bydureon as opposed to Metformin for T2D sufferers is I don’t want to produce more insulin. I want to let those little beta-cells rest up, and last for as long as possible.
Keep up the good work, my friend. And for those who have been diagnosed with RBD, get educated and get moving. Doing things that might help and can’t hurt are just common sense.
LikeLiked by 2 people
Hi Gavril,
Thanks for the interesting comment and sharing your information.
I am also with Diana – I think the way we currently conduct clinical trials is bonkers. Utterly nonsensical (https://scienceofparkinsons.com/2018/03/27/drug-trials/). I particularly don’t like folks being part of a placebo group for 12-24 months. But we need better and more objective methods of assessment in order to change this process.
I agree on the RBD and MSA angles, and I suspect that a clinical trial of a GLP-1 agonist in MSA is not too far away (https://www.multiplesystematrophy.org/blog/report-msa-coalition-funded-research-exendin-4).
I can not comment on your current treatment regime, but I would be curious to hear how you are determining if any of these changes are impacting your condition. What methods of self assessment are you using? We have a project brewing in the background here at the SoPD with regards to better self-assessment and we are looking for all the ideas we can find. More on this at a later date.
Kind regards,
Simon
LikeLike
If we’re speaking of possible RBD, my wife lets me know if I awaken her when I am moving in my sleep. She and I sleep very closely to one another, virtually in each other’s arms. She will touch me if I am moving to waken me if I am shaking. It’s always been around 4 in the morning during a dream, and started about 25 years ago during my divorce. (I really am going to have to have a formal video sleep study, because she also lets me know that I stop breathing rather often, and it frightens her.) I also had drenching night sweats, which have ceased.
If you read up on RBD, I seem to meet the criteria, but don’t know for sure. I also have essential tremor, for which I take a Russian atypical benzodiazipine called Gidazapam (Hydazepam?). Really works great, BTW. No addictive qualities, no hypnotic effects, just a relief from anxiety.
As I said, the isradipine causes fewer side effects than the old amlodipine, and while taking it, I’ve had no RBD (if that’s what they are) episodes. But then, add the Glycine, GABA, Melatonin, Selegiline, etc., and who can say?
From your site, I’ve learned way more about Parkinson’s than I hope I ever need to know, and have deep sympathy for those suffering with it.
LikeLike
Hi, I’m sorry and I hope not te be rude, but are you sure you have RBD ? I was sure that I had it during nearly a year, before finally having the opportunity to do a sleep study (waiting time for an appointement is very long, and yet I live in Paris, I cannot imagine the waiting time for a person living far from a big city or in a smaller country). My wife described me my moves like those of the RBD sleepers, not like classic nocturnal terrors, so I was afraid. Stress even made me develop symptoms close to those of Parkinson’s (like restless leg syndrome).
And great news, I learned that I move only during my deep sleep ! No RBD mentioned, my REM sleep is perfectly normal. My sleep is still strange (I’m trying some prescribed medication, like high doses of melatonin), but I don’t have anymore the other syndromes like restless leg, and I feel relieved.
Again, I don’t want to be rude with you, of course you (and your wife) know better than me how you sleep, but I think many among us fear for nothing about PD, especially since RBD is more publicized and well known (which is a good thing, but not so for hypochondriacs like me ! )
LikeLike
Hi Manual,
Rude you are not. In fact, you were very helpful to me. There is a rule somewhere I think that says, never diagnose yourself. RBD is a condition that is rare (1% of the population?) and there are other things that can be responsible for movement in one’s sleep.
So, I don’t plan to give this much more thought until I have an actual sleep study. I’m privately insured in Germany, so I should be able to get into a center as soon as I can take the time.
Again, much thanks!
Gav
LikeLike
Simon,Simon,
Thanks for such an interesting blog.
It seems to me that the evidence in favour of exenatide having neuroprotective properties is weak. Let me explain. We need to look at the graph “Reduction in motor scores in Exenatide”.
We see lines joining the plots. This makes us think that there’s continuous sampling. But, in fact there’s only six time points: 0, 12, 24, 36, 48 and 60 weeks. In the graph we see three sections:
* 0 and 12 weeks, a big decline in UPDRS motor scores. This could just as easily be explained by a short-term change in symptoms. (Perhaps, exenatide has dopamine agonist properties.)
* 12, 24, 36 and 48 weeks. the exenatide group’s results run parallel to the placebo group, albeit at a lower level. This could just as easily be explained by the short-term change continuing.
* 48 and 60 weeks, the injections are stopped, so the short-term symptom reductions are lost, the UPDRS scores rises (worsens) accordingly and ends up 2 points lower(out of about 30) than the placebo group’s score.
We must take into account the fact that the exenatide was taken in addition to the existing drug regimen of the participants. We need to check whether a similar graph would be found if, rather than exenatide, an extra dose of levodopa, for instance, had been given.
Clinical trials can gain confidence both by increasing the sample size and by increasing the frequency of sampling.
John
LikeLike
Hi John,
Glad you liked the post – thanks for the interesting comment. You are right that the evidence supporting any declaration of ‘neuroprotection’ in the Phase II clinical trial is weak at the moment. But the researchers who conducted that study have also been reluctant to make any such claim.
And you are right about the exisiting treatment regime issue. The investigators tried to manage the effect of the dopamine-based medication. But among the differences between the two groups in the study was that the total dose of Levodopa was lower in the Exenatide group at the start of the study compared to the placebo group (774mg vs 825mg, respectively), and the participants in the Exenatide group had greater increases in their dopaminergic therapy over the course of the trial (changes in current medication were allowed throughout the trial to minimise drop out from either group). So if the Exenatide group were increasing their Levodopa treatment over the course of the study and Exenatide was affecting the dopamine system, then perhaps the findings of the study could be explained by dopamine-based mechanisms.
Having said all of that, all of the clinical assessments were made during the off-medication state. In the report, off-medication was defined in the methods section of the report as “a period of withdrawal of levodopa for at least 8 h (ie, overnight) or 36 h in the case of long-acting drugs such as ropinirole, pramipexole, rasagiline, and rotigotine”.
I guess we need a bigger, longer phase III study in order to better determine exactly what (if anything) is happening.
Kind regards,
Simon
LikeLike
Simon, your blog brings incredible value to those of us with PD. Thank you for your tireless work.
I wanted to mention for those in the U.S. the Cedars Sinai study for a similar, possibly even superior, drug called Victoza. Info on the trial can be found at: https://www.cedars-sinai.edu/Patients/Programs-and-Services/Neurology/Clinical-Trials/Liraglutide-in-Parkinsons-Disease.aspx
I have spoken with the researchers there and here is what I learned: they are easy to work with and willing to take volunteers from anywhere in the U.S. as long as you can meet the requirements to be onsite.
Thanks again and thanks for all those individuals doing the heavy lifting for all of us by participating in a trial!
Pamela
LikeLiked by 1 person
Actually, he is saving lives through this blog God bless him and his work!
LikeLike
Hi Pamela,
Thanks for your comment and for the kind words about the website.
In addition to the Victoza (also known as Liraglutide) trial at Cedar Sinai (https://clinicaltrials.gov/ct2/show/NCT02953665) that you mentioned, there is also a phase II clinical trial of the GLP1-agonist Lixisenatide being conducted in France (https://clinicaltrials.gov/ct2/show/NCT03439943). Lots of GLP1-agonist research going on at the moment. Interesting times.
Kind regards,
Simon
LikeLike
Simon,
I keep on hearing that we do not know the longterm effects of exenatide for people with PD. BUT we do know the longterm effects without exenatide for people with PD (not great). And what about people with both diabetes and PD, are they not offered exenatide due to their PD? I thought repurposing of old drugs would be a quicker process…
Btw, thanks for all the great posts!
LikeLike
Hi Knas,
Glad you liked the post. You make a fair point. But the issue with clinical trials is that as you go further on along with the process, the bigger and more expensive the trials get. And the current bottleneck in the process is finding the resources to fund these larger & longer critical test trials.
I do know that there are a lot of efforts being made to investigate the long-term use of GLP1 agonists in the diabetic population. The goal is to determine if this particular class of drugs reduces the risk of people with diabetes to go on to develop PD. We should hear more about this before years end.
But yes, the clinical trial system is a slow process. We really need someone to propose a better method.
Simon
LikeLike
Simon
Another interesting, well written but cautious piece on a potential treatment for PD.
When will a treament be clinically available?
KW
LikeLiked by 1 person
Hi Peasant Farmer,
Thanks for the comment – I hope all is well. There are currently two phase II clinical trials for other GLP1 agonists ongoing in France and California at present and a Exenatide III trial is currently being planned. That phase III trial will most likely be conducted over 2-4 years, so 2023 is probably the best time horizon for Exenatide being approved for clinical use (if it is successful at the phase III stage).
NLY01 is on a fast track to human testing, with with the goal of a Phase 2 in Parkinson’s in 2019 (more information here: https://www.alzforum.org/news/research-news/does-taming-killer-astrocytes-spare-neurons-parkinsons-disease). But this will take longer than Exenatide.
Kind regards,
Simon
LikeLike
Hi Simon
Just a few thoughts from a Parkinsons patient – so apologies if not scientifically robust .
1)participants are described as being six years on average diagnosed – so they will still have pretty good store of naturally occurring dopamines – given this,the placebo effect is almost unavoidable – It’s a natural response which patients cant control -for a lot of more recently diagnosed patients , the placebo effect will be seen not only by thinking they are on a new drug but by being on a trial , in a hospital ,receiving attention from top doctors etc
The interesting thing is that if correctly randomised ,,the treatment group would also on average be 6 years diagnosed and would also undoubtedly be exhibiting the placebo effect (you can be a member of the treatment group and exhibit placebo )
The results chart show that the treatment group’s results are better than placebo groups – so therefore simply put – is the difference in scores between the two groups the difference Exenatide made ? (Taking out placebo effect at work in both groups )
( although – see below – after 2nd or 3rd test i would think the improvement made is mainly due to drug )
2)one of the things ,which really frustrates me about the whole placebo thing -is that , the very group of people , namely those with 6-10 years of diagnosis , who would most benefit from a treatment , will ,with their naturally occurring dopamines be very likely to exhibit placebo – so should they be excluded from trials? – of course not – we need to somehow better cater for the placebo effect and not automatically assume that if seen , it , in some way invalidates the treatment . In shorter term diagnosed patients I believe they almost fall into a group along with with non sufferers , who when they believe they are on to something good or beneficial will exhibit ‘placebo ‘ However, if the individuals are not rewarded within an acceptable timeframe the placebo effect will wear off . This can be seen in the results of the trial with the placebo group scores gradually getting worse,as the initial euphoria wears thin and the control group realises there will be no ‘prize’ for them – they are on the placebo !
3) conversely were there any longer term patients like me (17 years ) on this trial – i had no placebo effect at all on my trial due to health being so bad and dopamine levels so low that something particularly spectacular has to happen for it to kick into action (like finding that perfect outfit in TK Maxx for £10 !) Seriously ,a treatment trial is such an ordeal for longer term sufferers that the placebo effect I’m guesssing would be pretty low and would definitely disappear after 2-3 UPDRS tests off drugs (horrendous ) Does any data analysis exist on length of diagnosis and placebo effect – from this trial or any other ?
An additional thought – as participants only on average 6 years diagnosed were there any issues with the ceiling effect or nil scores on UPDRS test ?
4) prior knowledge of Exenatide and it being purported as a ‘ wonder drug ‘ will definitely have influenced participants – it definitely happened with GDNF – but as i say we have to engage common sense and make sure valuable treatments are not lost by digging into the data.
5) the increasing scores post treatment also are what I’d expect a) because placebo results getting worse as realisation dawns so gap between groups widens b) levadopa and GDNF – not in all patients but many – start to work after being ‘ layered thinly on “ so results can take time
6) some quick ideas to stimulate debate about placebo. -1) in first two to three tests, when placebo at height, should these results be discounted ? 2) ask each patient every month whether on placebo or treatment – then at trial end we can determine effect placebo had on trial – ie if 100% of participants guessed correctly they were on placebo or treatment there is no evidence to suggest placebo at work , so results do not need to be adjusted in any way 3) once patient knows they are on placebo should we switch to treatment , as game is up so to speak ??? This would help with ethical concerns about time on placebo especially if patient knows . Or perhaps we could test PD treatments in another way which means a placebo or control group would not be required – I’m still working on that !
Regards
Carol
Ps Sorry its turned into a bit more than a comment !
LikeLike
I can tell you exenatide works. Whether it masks symptoms or is neuroprotective I cannot definitively say. What I can say is I have been on exenatide for nine months, since thanksgiving 2017. I used to have acting out of my dreams regularly (not every night, but regularly). I have not had one since that time.
LikeLike